Finding information about genes and variants in Ensembl, Demo
Ensembl homepage
The front page of Ensembl is found at ensembl.org. It contains lots of information and links to help you navigate Ensembl:
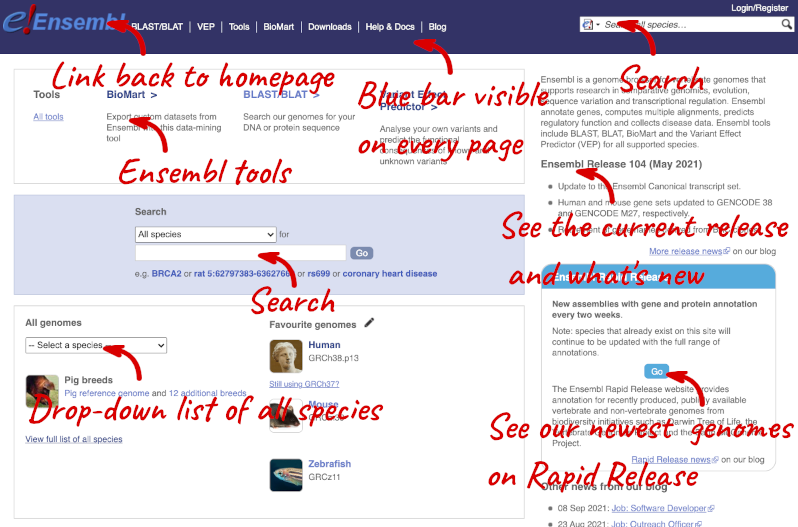
At the top left you can see the current release number and what has come out in this release. To access old releases, scroll to the bottom of the page and click on View in archive site.
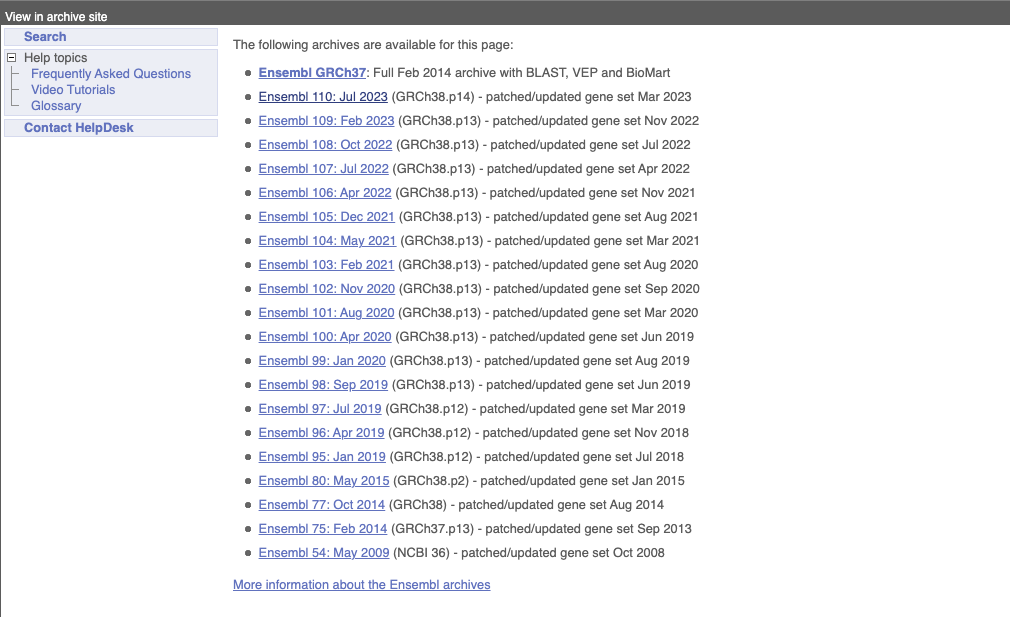
Click on the links to go to the archives. Alternatively, you can jump quickly to the correct release by putting it into the URL, for example e98.ensembl.org jumps to release 98.
Click on View full list of all species.
Click on the common name of your species of interest to go to the species homepage. We’ll click on Human.
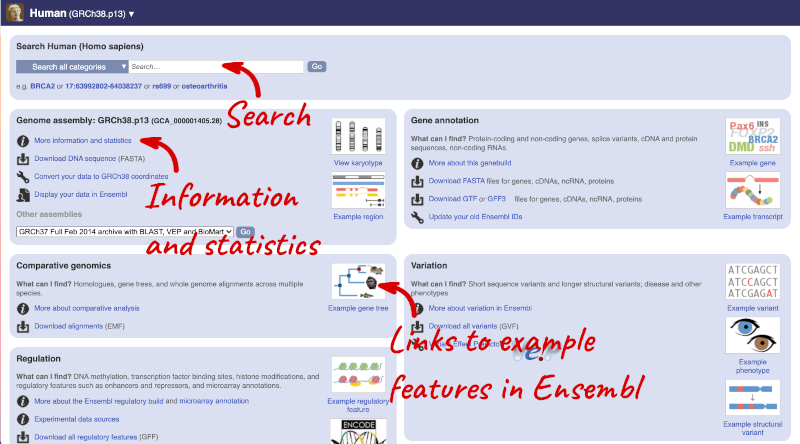
Here you can see links to example pages and to download flatfiles. To find out more about the genome assembly and genebuild, click on More information and statistics.
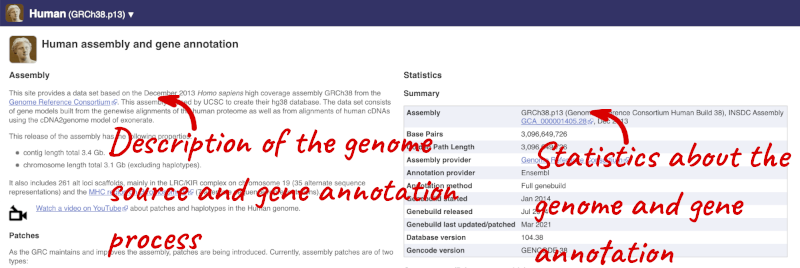
Here you’ll find a detailed description of how to the genome was produced and links to the original source. You will also see details of how the genes were annotated.
The current genome assembly for human is GRCh38. If you want to see the previous assembly, GRCh37, visit our dedicated site grch37.ensembl.org.
The gene tab
You can find out lots of information about Ensembl genes and transcripts using the browser. You can search for gene names or identifiers, and also phenotypes or functions that might be associated with the genes.
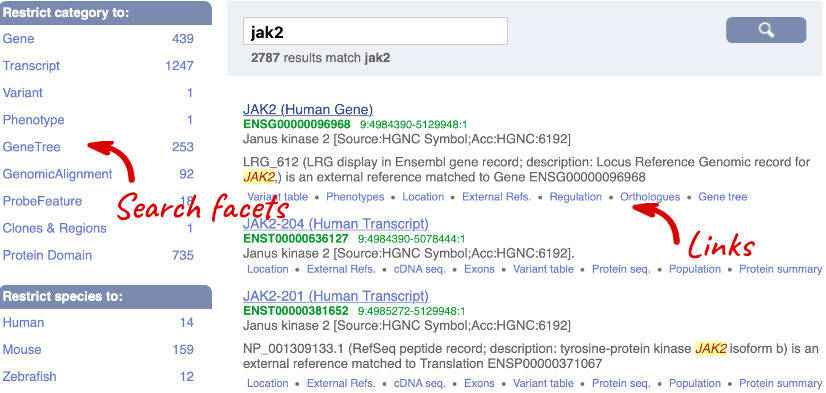
We’re going to look at the human JAK2 gene. From ensembl.org, type JAK2 into the search bar and click the Go button. You will get a list of hits with the human gene at the top.
Where you search for something without specifying the species, or where the ID is not restricted to a single species, the most popular species will appear first, in this case, human, mouse and zebrafish appear first. You can restrict your query to species or features of interest using the options on the left.
Click on the gene name or Ensembl ID. The Gene tab should open:
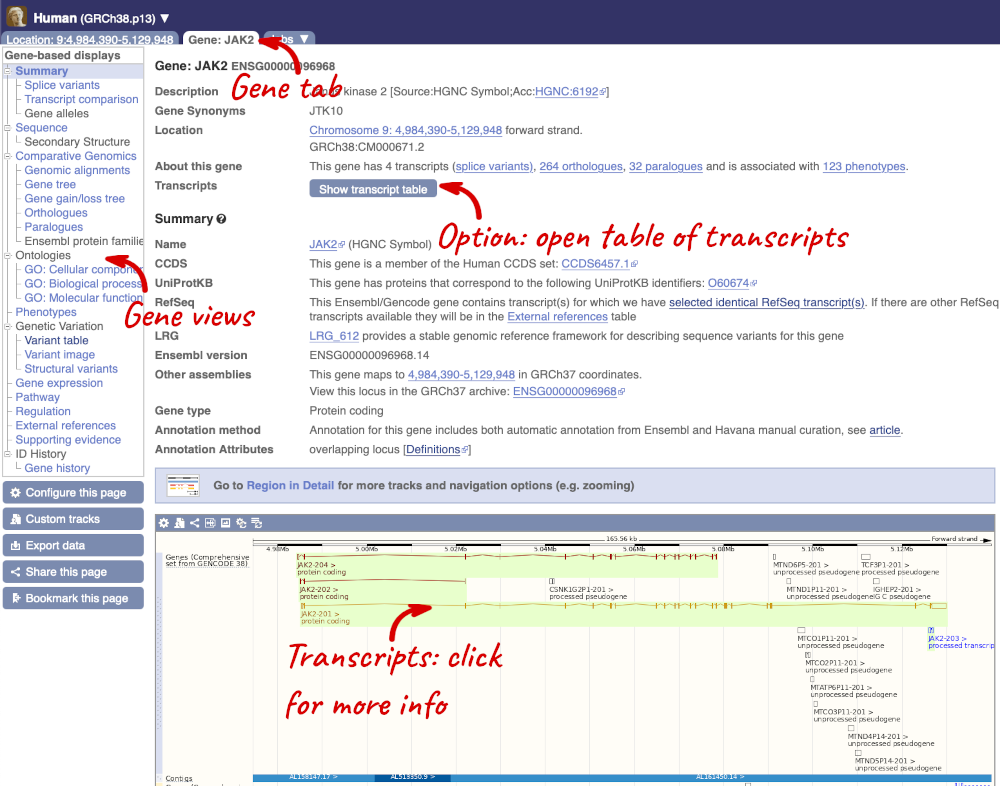
This page summarises the gene, including its location, name and equivalents in other databases. At the bottom of the page, a graphic shows a region view with the transcripts. We can see exons shown as blocks with introns as lines linking them together. Coding exons are filled, whereas non-coding exons are empty. We can also see the overlapping and neighbouring genes and other genomic features.
There are different tabs for different types of features, such as genes, transcripts or variants. These appear side-by-side across the blue bar, allowing you to jump back and forth between features of interest. Each tab has its own navigation column down the left hand side of the page, listing all the things you can see for this feature.
Let’s walk through this menu for the gene tab. How can we view the genomic sequence? Click Sequence at the left of the page.
The sequence is shown in FASTA format. The FASTA header contains the genome assembly, chromosome, coordinates and strand (1 or -1) – this gene is on the positive strand.

Exons are highlighted within the genomic sequence, both exons of our gene of interest and any neighbouring or overlapping gene. By default, 600 bases are shown up and downstream of the gene. We can make changes to how this sequence appears with the blue Configure this page button found at the left. This allows us to change the flanking regions, add variants, add line numbering and more. Click on it now.
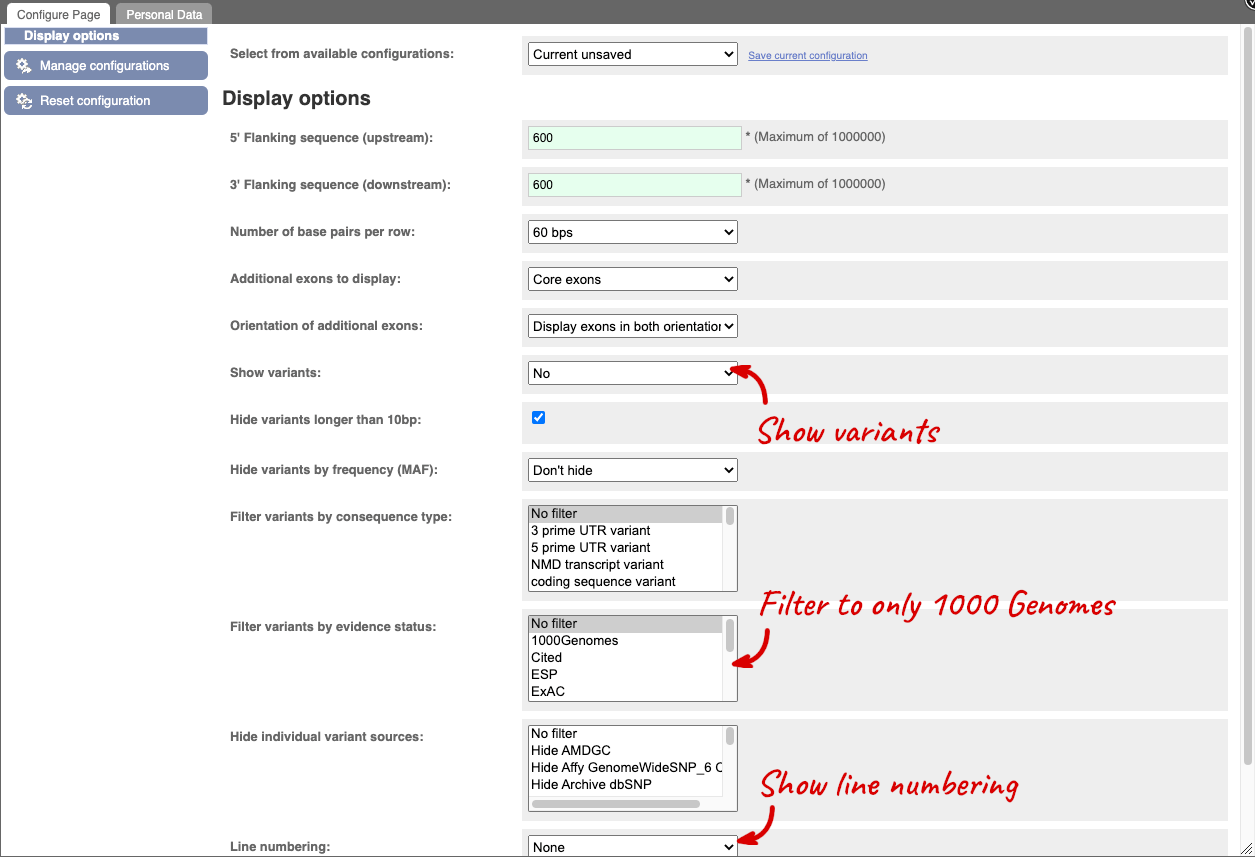
You can view variants on the sequence in any of the sequence views. Once you have selected changes (in this example, Show variants: Yes and show links, Filter variants by evidence status: 1000Genomes and Line numbering: Relative to coordinate system) click at the top right.
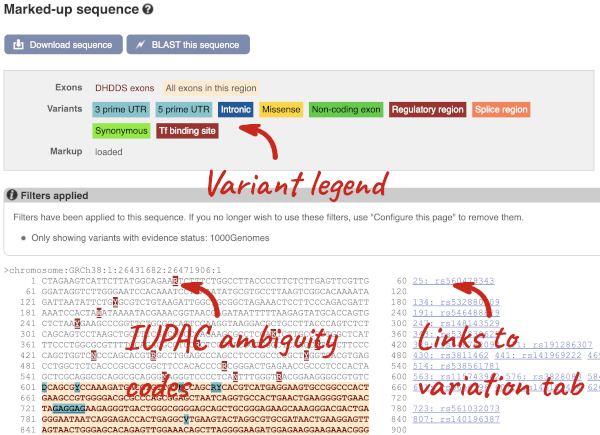
Find out more about a variant by clicking on it.
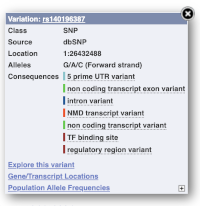
You can go to the Variation tab by clicking on the variant ID. For now, we’ll explore more ways of finding variants.
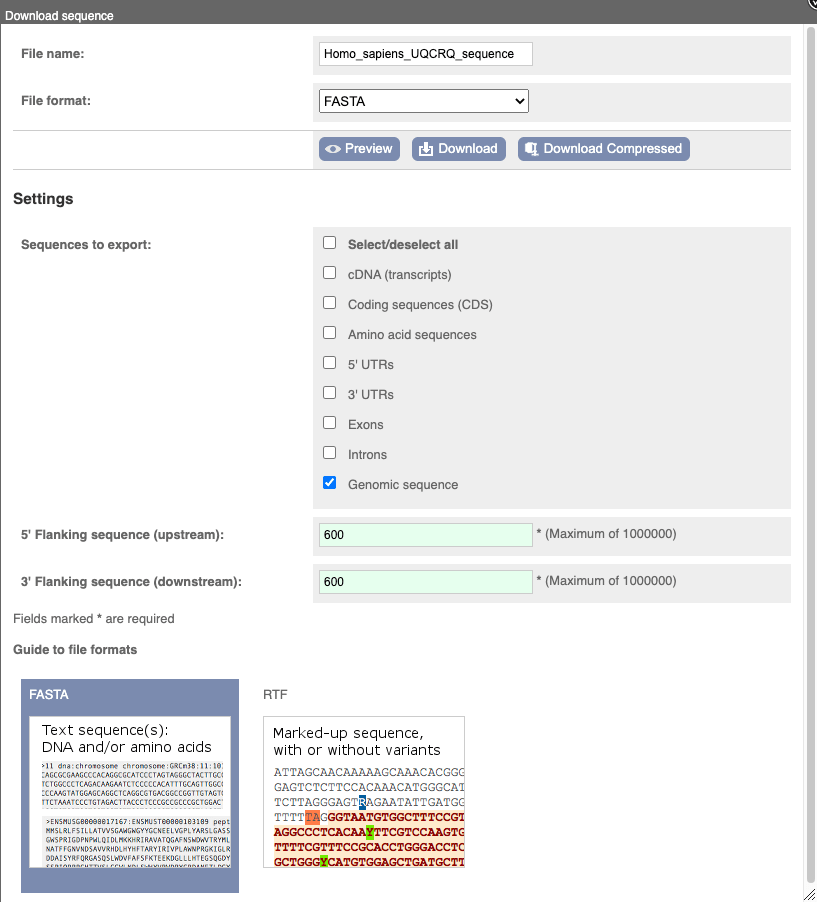
You can download this sequence by clicking in the Download sequence button above the sequence:

This will open a dialogue box that allows you to pick between plain FASTA sequence, or sequence in RTF, which includes all the coloured annotations and can be opened in a word processor. If you want run a sequence analysis tool, download as FASTA sequence, whereas if you want to analyse the sequence visually, RTF is best for this. This button is available for all sequence views.
To view all the sequence variants in table form, click the Variant table link at the left of the gene tab.
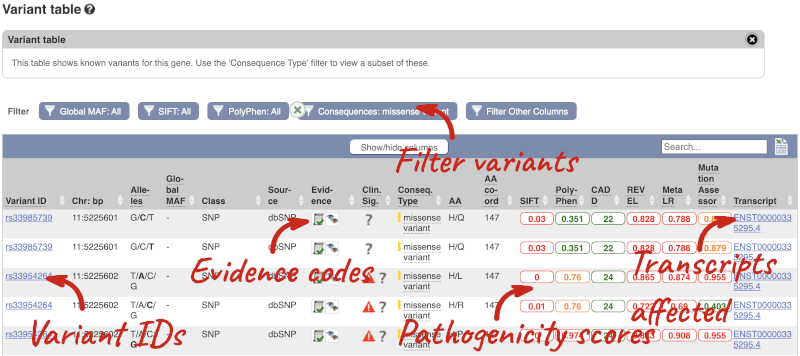
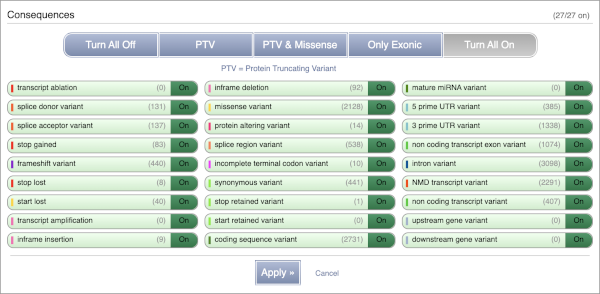
You can filter the table to only show the variants you’re interested in. For example, click on Consequences: All, then select the variant consequences you’re interested in. For display purposes, the table above has already been filtered to only show missense variants.
You can also filter by the different pathogenicity scores and MAF, or click on Filter other columns for filtering by other columns such as Evidence or Class.
The table contains lots of information about the variants. You can click on the IDs here to go to the Variation tab too.
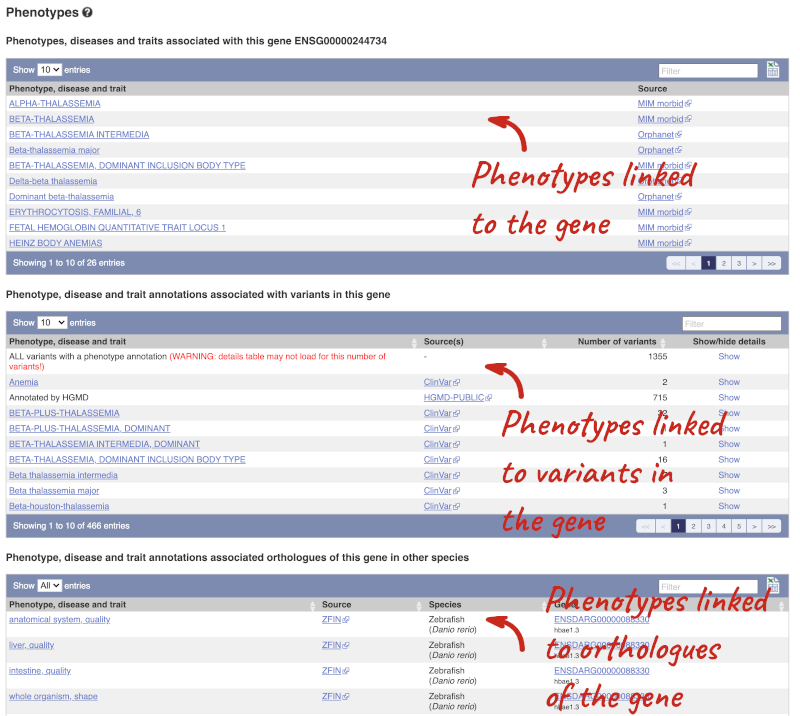
You can also see the phenotypes associated with a gene. Click on Phenotype in the left hand menu.
The transcript tab
We’re now going to explore the different transcripts of JAK2. Click on Show transcript table at the top.
![]()
![]()
Here we can see a list of all the transcripts of JAK2 with their identifiers, lengths, biotypes and flags to help you decide which ones to look at.
If we were to only choose one transcript to analyse, we would choose JAK2-201 because it is the MANE Select and Ensembl Canonical. This means it is both 100% identical to the RefSeq transcript NM_004972.4 and both Ensembl and NCBI agree that it is the most biologically important transcript.
Click on the ID, ENST00000381652.4.
You are now in the Transcript tab for JAK2-201. We can still see the gene tab so we can easily jump back. The left hand navigation column provides several options for the transcript JAK2-201 - many of these are similar to the options you see in the gene tab, but not all of them. If you can’t find the thing you’re looking for, often the solution is to switch tabs.
Click on the Exons link. This page is useful for designing RT-PCR primers because you can see the sequences of the different exons and their lengths.
![]()
You may want to change the display (for example, to show more flanking sequence, or to show full introns). In order to do so click on Configure this page and change the display options accordingly.
Now click on the cDNA link to see the spliced transcript sequence with the amino acid sequence. This page is useful for mapping between the RNA and protein sequences, particularly genetic variants.
![]()
UnTranslated Regions (UTRs) are highlighted in dark yellow, codons are highlighted in light yellow, and exon sequence is shown in black or blue letters to show exon divides. Sequence variants are represented by highlighted nucleotides and clickable IUPAC codes are above the sequence.
Click on Haplotypes in the left hand menu.
![]()
The Haplotypes view in the transcript tab shows you the actual protein and CDS sequences in 1000 Genomes individuals. This is possible because the 1000 Genomes study has phased genotypes, so we know which alleles occur on which of the chromosome pairs. The table lists all the versions of the protein that occur along with their frequencies, including the reference sequence and sequences with one or more alternative alleles.
Click on one of the haplotypes, we’ll go for 393L>V, to find out more about it. Here you will see the frequency in the 1000 Genomes subpopulations, the sequence and the 1000 Genomes individuals where this protein is found.
The variant tab
Blood cancers known as MPNs commonly occur due to an activating somatic mutation (JAK2-V617F) in the JAK2 gene decades before diagnosis. Let’s have a look at a specific variant. We could find the variant COSV67569051 in the Variant table, however it’s easier to find if we put COSV67569051 into the search box. Click through to open the Variation tab.
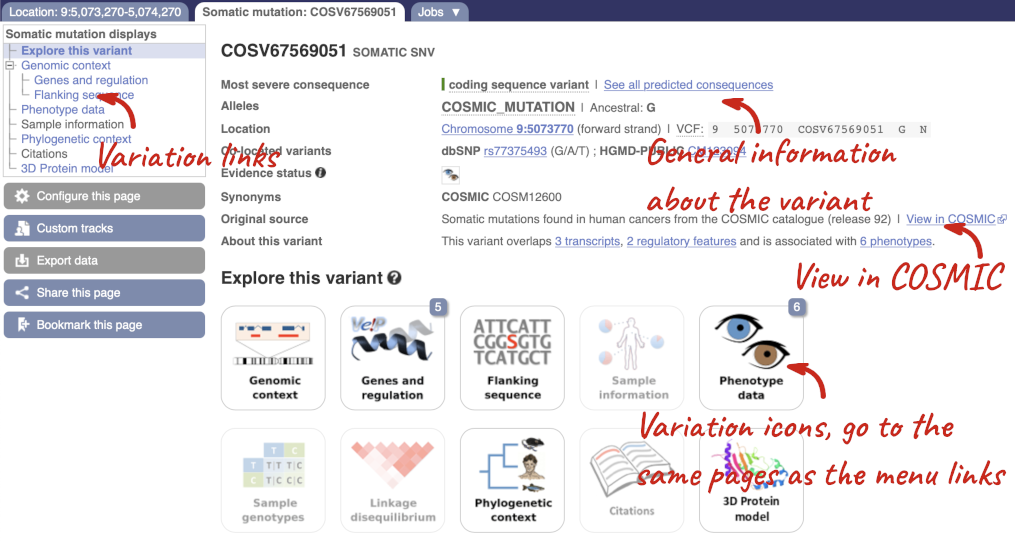
The icons show you what information is available for this variant. Click on Genes and regulation, or follow the link on the left.
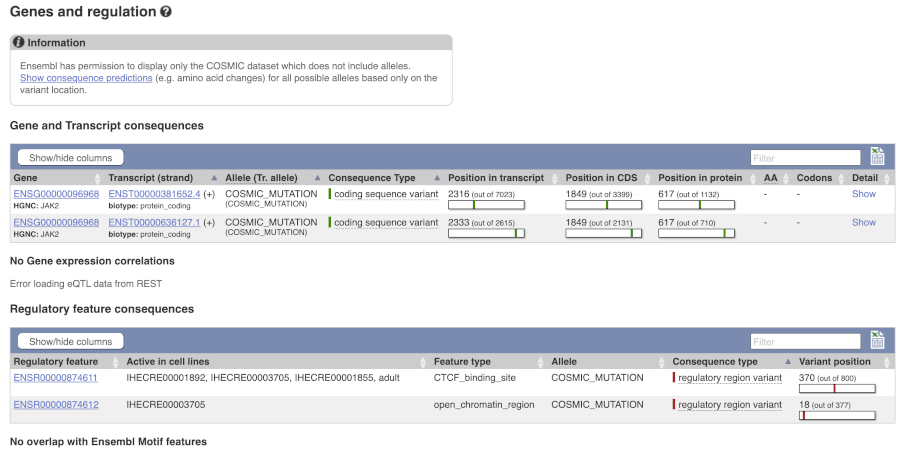
We can see the variant falls in a coding region, as the consequence is coding sequence variant. Ensembl cannot display the alleles for COSMIC variants, so you will need go to the COSMIC database to find out if it is a missense or synonymous mutation. Click on View in COSMIC in the panel above, and then c.1849G>T to go to the variant page.
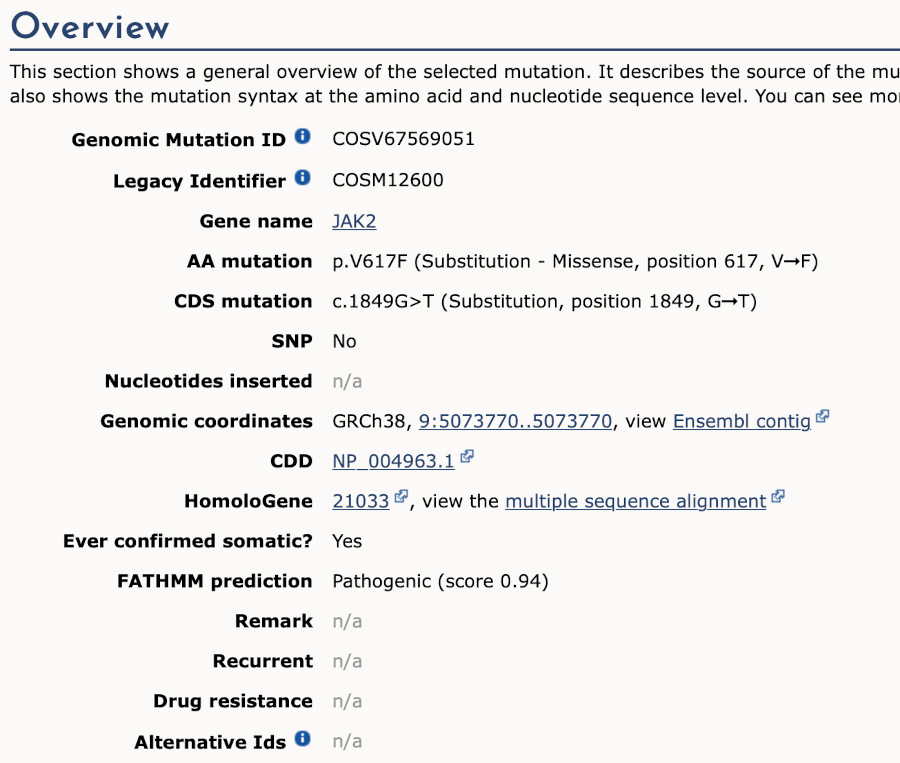
We now know that this variant alleles are G>T, causing V617F amino acid substitution. Go back to the variant tab in Ensembl and click on dbSNP variant rs77375493 under Co-located variants in the overview panel.

It is the same variant, but imported from dbSNP. We can see that there is more information for this dbSNP variant, compared to the COSMIC variant. We can see the consequence, alternate alleles and allele frequency data. Let’s click on Genes and regulation now.
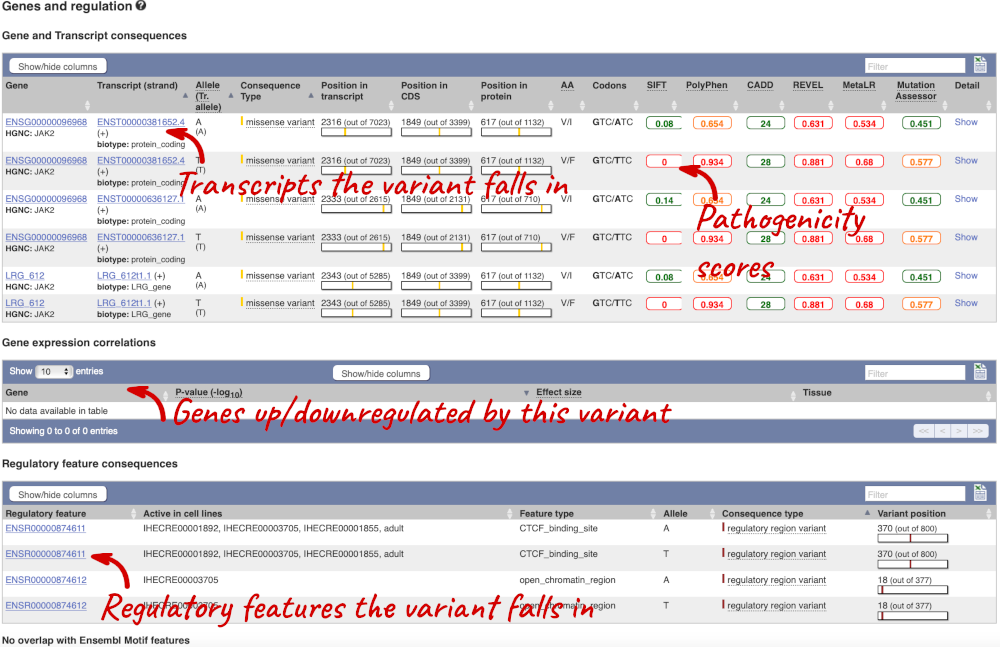
This page illustrates the genes the variant falls within and the consequences on those genes, including pathogenicity predictors, but also regulatory features that the variant falls within.
Let’s look at population genetics. Click on Population genetics in the left-hand menu.
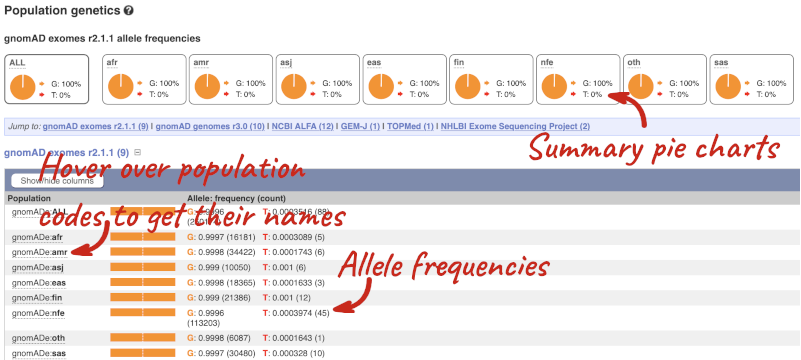
This variant is very rare and has not been found in 1000 Genomes populations, but has been reported in some diseased cohorts. The population allele frequencies are shown by study, including gnomAD, NCBI ALPHA, GEM-J, TOPMed and NHLBI Exome Sequencing Project. Where genotype frequencies are available, these are shown in the tables.
Let’s have a look at the phenotypes associated with this variant. Click on Phenotype Data in the left-hand menu.
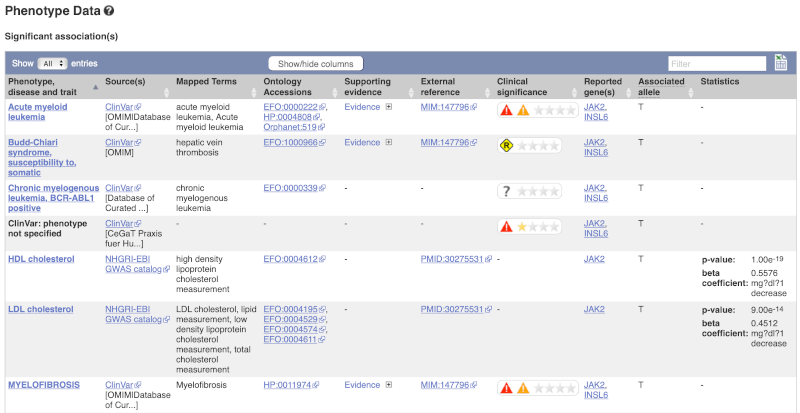
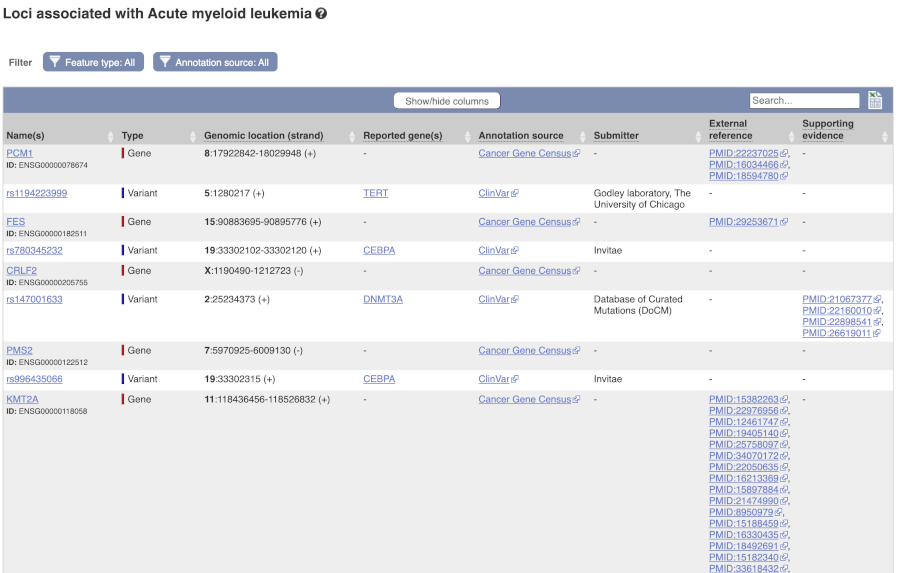
This variant is associated with various phenotypes, including blood cancers such as acute myeloid leukemia, myelofibrosis and increased numbers of mature blood cells. These phenotype associations come from sources including the GWAS catalog, ClinVar, Orphanet and OMIM. Where available, the table provides links to the original paper that made the association, the allele that is associated with the phenotype and p-values and other statistics.
Are there other variants in the genome that also cause acute myeloid leukemia? Click on the phenotype Acute myeloid leukemia to find out.