Filter Events by Year
Ensembl VEP Command Line Tutorial - Genome Bioinformatics 2024
Course Details
- Lead Trainer
- Jorge Batista da Rocha
- Event Date
- 2024-11-20
- Location
- Genome bioinformatics: from short- to long-read sequencing 2024 - EMBL-EBI, Wellcome Genome Campus, Hinxton, UK
- Description
- Work with the Ensembl Outreach team to practice using the Variant Effect Predictor (VEP) in command line format.
Demos and exercises
VEP cmd GenBionf2024
Command-line VEP analysis of variants from a 1000 Genomes Project dataset
Ensembl’s Variant Effect Predictor (VEP) is a powerful tool for annotating genomic variants. VEP is accessible via web, REST API and command line options.
In this practical session, we will practice using VEP via command line to annotate a variant call file (VCF).
The VCF you will use contains variant calls for Homo Sapiens chromosome 13 from the IGSR: The International Genome Sample Resource. This file was extracted using Ensembl’s Data Slicer for human variation aligned to GRCh38, with the IGSR British in England and Scotland GBR population samples subsetted.
Directories and starting tutorials:
A general tutorial for command line VEP is available to try out or compare to if you need some guidance.
VEP is installed in the /home/training/ensembl-vep directory. Change to this directory to complete the exercises below.
The subsetted IGSR VCF file name is 13.32315086-32400268.ALL.chr13_GRCh38.genotypes.20170504.GBR.vcf
Exercises:
-
Explore the VCF file with any text viewer to explore its contents. What do lines denoted by “##” represent? What are the key headers in the file? You use VCF file specifications as a reminder.
-
VEP is installed in the
/home/training/ensembl-vepUse the command-line VEP tool annotate the variants in the IGSR VCF file and output to VCF format. Here is an example command./vep -i examples/homo_sapiens_GRCh38.vcf --cache --vcf -o example_homo_sapiens_GRCh38output.vcf -
Explore the output of VEP in any text viewer to explore the contents. What genes are affected by the variants in the file?
-
VEP also produces an HTML output file - try exploring this file with a browser tool such as Firefox or Chrome. How many variants were annotated? What is the proportion of variants that are of the consequence “missense_variant”?
-
Re-run the command-line VEP tool to annotate the variants in your VCF including if they occur in a MANE and Ensembl Canonical transcript. Save the output of this query into a separate output file in the default text format (omit –vcf).
-
Use the
filter_VEPtool to find variants that are located within the BRCA2 gene in a MANE transcript. Are there any missense variants present in this filter? -
If you are done with the above, you may try the Web version of VEP on your file by uploading the VCF. Try the different options available and see how these can add different layers of information. Also note the command used in the job completion page as this can help you when adapting to command line!
- You can open the VCF file with gedit, or by using a command such as:
less -S 13.32315086-32400268.ALL.chr13_GRCh38.genotypes.20170504.GBR.vcfthen use the arrow keys to navigate, and press Q when ready to quit. Lines starting with ## indicate information headers on how the file was processed.
- You can run VEP on the VCF input file using the following script:
./vep -i 13.32315086-32400268.ALL.chr13_GRCh38.genotypes.20170504.GBR.vcf -o VEP_annot_chr13_GBR.vcf --vcf --cache
Your own script may not look exactly like this and you may employ different flags:
--input_file or -i Allows you to specify the location of the input file. --output_file or -o Allows you to specify the name of the output file.
--force_overwrite Allows VEP to overwrite a pre-existing output file with the same name.
--genomes Points VEP to the Ensembl Genomes (non-vertebrates) server. (not used for this homo sapiens example)
--cache Enables the use of the cache (this can speed up VEP significantly).
--cache_verson Allows you to specify the cache version. This should be used with Ensembl Genomes caches, since their version numbers do not match Ensembl versions. For example, the VEP/Ensembl version may be 110 and the Ensembl Genomes version 57.
--check_existing Checks for the existence of known variants that are co-located with your input variants.
--offline Enables offline mode (no database connections are made).
View the output for exercise 1 here.
- You may use a similar less command or method to open the annotated VCF file:
less -S VEP_annot_chr13_GBR.vcfthere are two genes listed in the annotations, ZAR1L ENSG00000189167 and BRCA2 ENSG00000139618
- Open the HTML file by selecting to open it with a browser, or directing a browser command to it:
firefox VEP_annot_chr13_GBR.vcf_summary.htmlOnly 1.1% of variants are missense variants View the output for exercise 4 here.
- Rerun VEP with the following command
./vep -i 13.32315086-32400268.ALL.chr13_GRCh38.genotypes.20170504.GBR.vcf -o VEP_annot_chr13_GBR_MANE_Can.txt --canonical --mane --cache --symbol
View the output for exercise 5 here.
- Filter for BRCA2 and MANE with the following command
filter_vep -i VEP_annot_chr13_GBR_MANE_Can.txt -o VEP_annot_chr13_GBR_MANE_Can_filt_BRCA_MANE.txt --filter "SYMBOL is BRCA2 and MANE_SELECT exists"View output for exercise 6 here There is a missense variant - rs80358762 - you can explore phenotype information related to that variant here
web VEP
We have identified five variants on human chromosome nine, C-> A at 128203516, an A deletion at 128328461, C->A at 128322349, C->G at 128323079 and G->A at 128322917.
We will use the Ensembl VEP to determine:
- Have my variants already been annotated in Ensembl?
- What genes are affected by my variants?
- Do any of my variants affect gene regulation?
Go to the front page of Ensembl and click on the Variant Effect Predictor.

This page contains information about the VEP, including links to download the script version of the tool. Click on Launch VEP to open the input form:
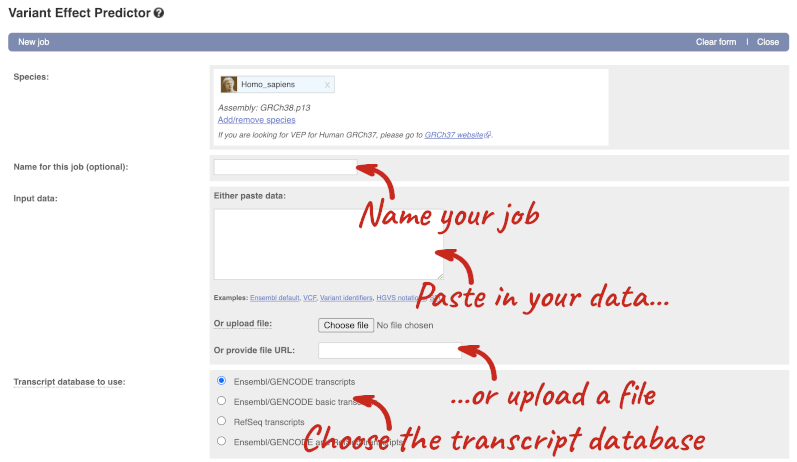
The data is in VCF format:
chromosome coordinate id reference alternative
Put the following into the Paste data box:
9 128328460 var1 TA T
9 128322349 var2 C A
9 128323079 var3 C G
9 128322917 var4 G A
9 128203516 var5 C A
The VEP will automatically detect that the data is in VCF.
There are further options that you can choose for your output. These are categorised as Identifiers, Variants and frequency data, Additional annotations, Predictions, Filtering options and Advanced options. Let’s open all the menus and take a look.
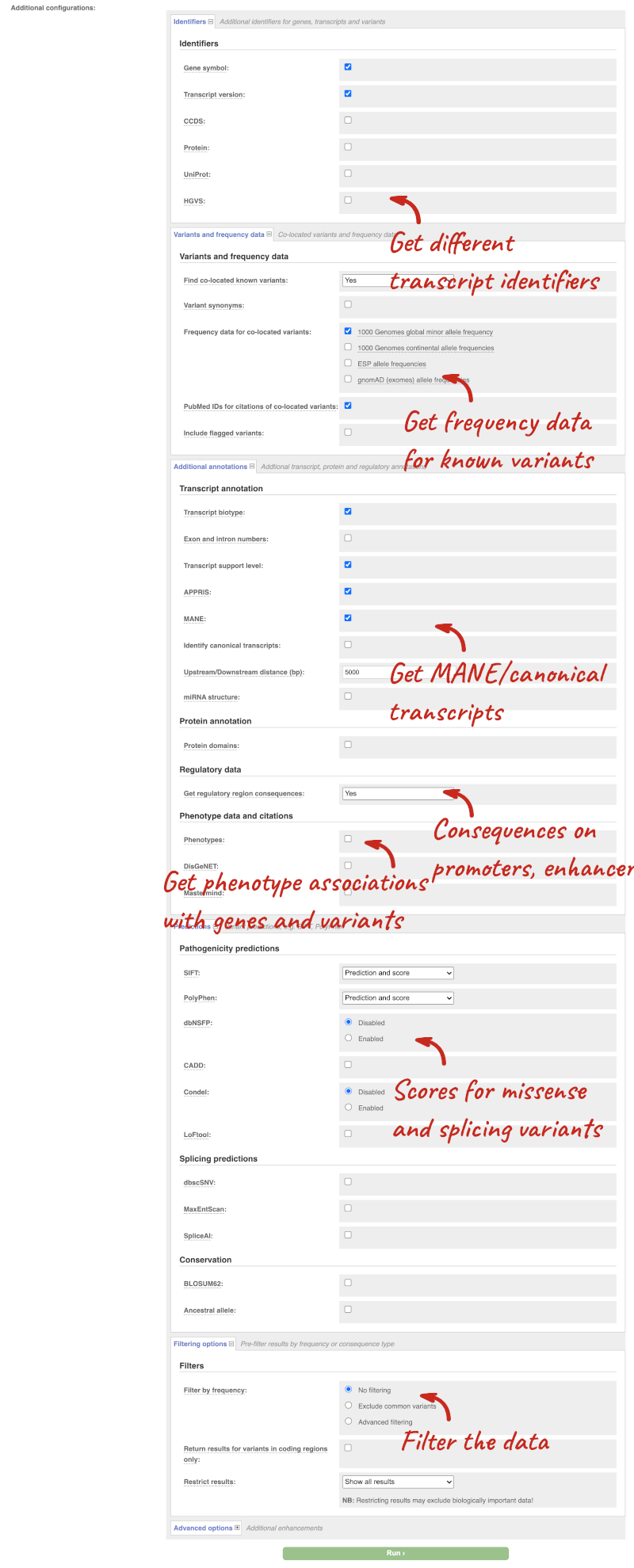
Hover over the options to see definitions.
We’re going to select some options:
- HGVS, annotation of variants in terms of the transcripts and proteins they affect, commonly-used by the clinical community
- Phenotypes
- Protein domains
When you’ve selected everything you need, scroll right to the bottom and click Run.

The display will show you the status of your job. It will say Queued, then automatically switch to Done when the job is done, you do not need to refresh the page. You can edit or discard your job at this time. If you have submitted multiple jobs, they will all appear here.
Click View results once your job is done.
In your results you will see a graphical summary of your data, as well as a table of your results.
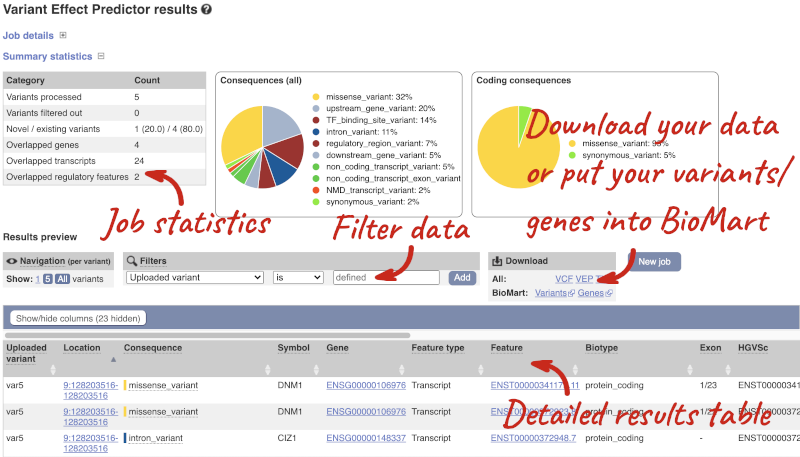
The results table is enormous and detailed, so we’re going to go through the it by section. The first column is Uploaded variant. If your input data contains IDs, like ours does, the ID is listed here. If your input data is only loci, this column will contain the locus and alleles of the variant. You’ll notice that the variants are not neccessarily in the order they were in in your input. You’ll also see that there are multiple lines in the table for each variant, with each line representing one transcript or other feature the variant affects.
You can mouse over any column name to get a definition of what is shown.
The next few columns give the information about the feature the variant affects, including the consequence. Where the feature is a transcript, you will see the gene symbol and stable ID and the transcript stable ID and biotype. Where the feature is a regulatory feature, you will get the stable ID and type. For a transcription factor binding motif (labelled as a MotifFeature) you will see just the ID. Most of the IDs are links to take you to the gene, transcript or regulatory feature homepage.
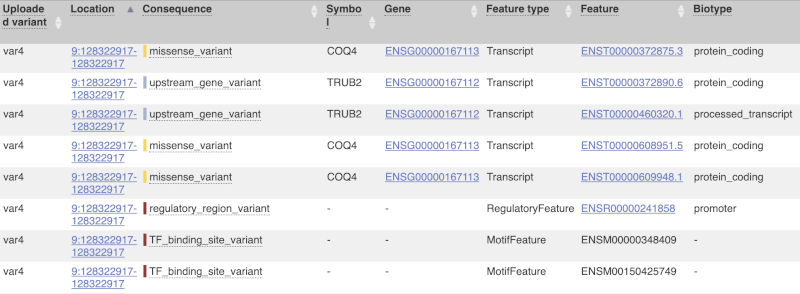
This is followed by details on the effects on transcripts, including the position of the variant in terms of the exon number, cDNA, CDS and protein, the amino acid and codon change, transcript flags, such as MANE, on the transcript, which can be used to choose a single transcript for variant reporting, and predicted pathogenicity scores. The predicted pathogenicity scores are shown as numbers with coloured highlights to indicate the prediction, and you can mouse-over the scores to get the prediction in words. Two options that we selected in the input form are the HGVS notation, which is shown to the left of the image below and can be used for reporting, and the Domains to the right. The Domains list the proteins domains found, and where there is available, provide a link to the 3D protein model which will launch a LiteMol viewer, highlighting the variant position.
![]()
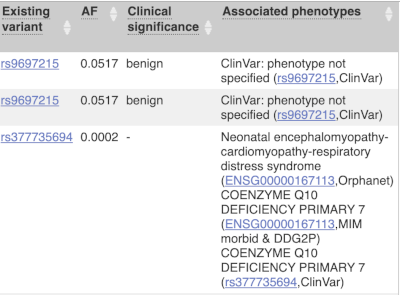
Where the variant is known, the ID of the existing variant is listed, with a link out to the variant homepage. In this example, only rsIDs from dbSNP are shown, but sometimes you will see IDs from other sources such as COSMIC. The VEP also looks up the variant from frequency files, and pulls back the allele frequency (AF in the table), which will give you the 1000 Genomes Global Allele Frequency. In our query, we have not selected allele frequencies from the continental 1000 Genomes populations or from gnomAD, but these could also be shown here. We can also see ClinVar clinical significance and the phenotypes associated with known variants or with the genes affected by the variants, with the variant ID listed for variant associations and the gene ID listed for gene associations, along with the source of the association.
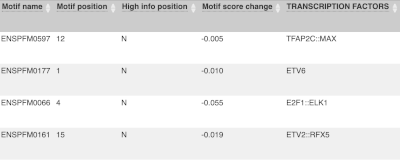
For variants that affect transcription factor binding motifs, there are columns that show the effect on motifs (you may need to click on Show/hide columns at the top left of the table to display these). Here you can see the position of the variant in the motif, if the change increases or decreases the binding affinity of the motif and the transcription factors that bind the motif.
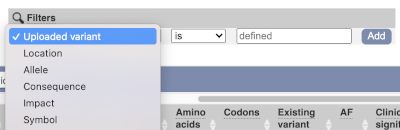
Above the table is the Filter option, which allows you to filter by any column in the table. You can select a column from the drop-down, then a logic option from the next drop-down, then type in your filter to the following box. We’ll try a filter of Consequence, followed by is then missense_variant, which will give us only variants that change the amino acid sequence of the protein. You’ll notice that as you type missense_variant, the VEP will make suggestions for an autocomplete.
You can export your VEP results in various formats, including VCF. When you export as VCF, you’ll get all the VEP annotation listed under CSQ in the INFO column. After filtering your data, you’ll see that you have the option to export only the filtered data. You can also drop all the genes you’ve found into the Gene BioMart, or all the known variants into the Variation BioMart to export further information about them.
Running CFTR variants through VEP
Resequencing of the genomic region of the human CFTR (cystic fibrosis transmembrane conductance regulator (ATP-binding cassette sub-family C, member 7) gene (ENSG00000001626) has revealed the following variants. The alleles defined in the forward strand:
- G/A at 7: 117,530,985
- T/C at 7: 117,531,038
- T/C at 7: 117,531,068
Use the VEP tool in Ensembl and choose the options to see SIFT and PolyPhen predictions. Do these variants result in a change in the proteins encoded by any of the Ensembl genes? Which gene? Have the variants already been found?
Go to the Ensembl homepage and click on the link Tools at the top of the page. Currently there are nine tools listed in that page. Click on Variant Effect Predictor and enter the three variants as below:
7 117530985 117530985 G/A
7 117531038 117531038 T/C
7 117531068 117531068 T/C
Note: Variation data input can be done in a variety of formats. See more details about the different data formats and their structure in this VEP documentation page. Click Run. When your job is listed as Done, click View Results.
You will get a table with the consequence terms from the Sequence Ontology project (http://www.sequenceontology.org/) (i.e. synonymous, missense, downstream, intronic, 5’ UTR, 3’ UTR, etc) provided by VEP for the listed SNPs. You can also upload the VEP results as a track and view them on Location pages in Ensembl. SIFT and PolyPhen are available for missense SNPs only. For two of the entered positions, the variations have been predicted to have missense consequences of various pathogenicity (coordinate 117531038 and 117531068), both affecting CFTR. All the three variants have been already annotated and are known as rs1800077, rs1800078 and rs35516286 in dbSNP (databases, literature, etc).
VEP analysis of structural variants in human
We have details of a genomic deletion in a breast cancer sample in VCF format:
13 32307062 sv1 . <DEL> . . SVTYPE=DEL;END=32908738
Use VEP in Ensembl to find out the following information:
1. How many genes have been affected?
2. Does the structural variant (SV) cause deletion of any complete transcripts?
3. Map your variant in the Ensembl browser on the Region in detail view.
- Click on VEP at the top of any Ensembl page and open the web interface. Make sure your species is Human. It is good practise to name your VEP jobs something descriptive, such as Patient deletion exercise. Paste the variant in VCF format into the Paste data field and hit Run.
12 different genes are affected by the SV.
- Filter your table by select Consequence is
transcript_ablationat the top of the table and click Add.Yes, there is deletion of complete transcripts of PDS5B, N4BP2L1, BRCA2, RNY1P4, IFIT1P1, ATP8A2P2, N4BP2L2, N4BP2L2-IT2 and one gene without official symbols: ENSG00000212293.
- To view your variant in the browser click on the location link in the results table 13: 32307062-32908738. The link will open the Region in detail view in a new tab. If you have given your data a name it will appear automatically in red. If not, you may need to Configure this page and add it under the Personal data tab in the pop-up menu.