Filter Events by Year
CABANA Workshop: Plant bioinformatics - analysis of crop genomics data, 13-17 December 2021
Course Details
- Lead Trainer
- Ben Moore
- Associate Trainer(s)
- Event Dates
- 2021-12-13 until 2021-12-17
- Location
- Virtual
- Description
- Work with the Ensembl Outreach team to get to grips with the Ensembl Plants browser, accessing gene, variation and comparative genomics data.
Demos and exercises
Ensembl Plants genes and transcripts
The front page of Ensembl Plants is found at plants.ensembl.org. It contains lots of information and links to help you navigate Ensembl Plants:
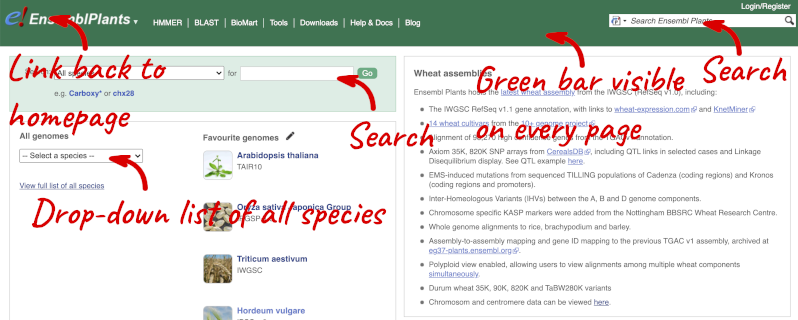
At the top left you can see the current release number and what has come out in this release.
Click on View full list of all species.
Click on the common name of your species of interest to go to the species homepage. We’ll click on Triticum aestivum.
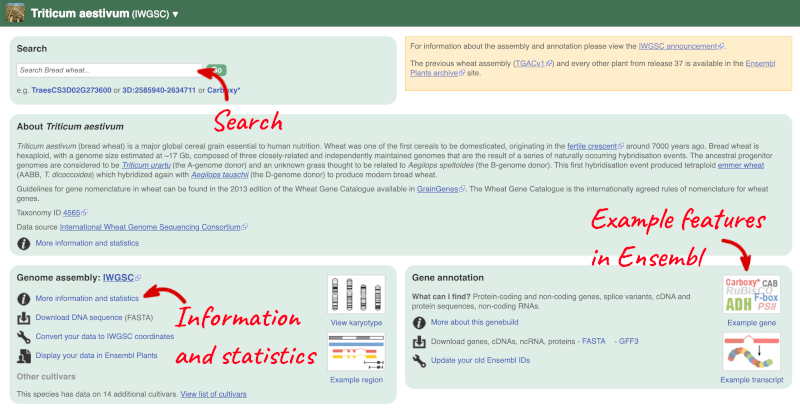
Here you can see links to example pages and to download flatfiles. To find out more about the genome assembly and genebuild, click on More information and statistics.
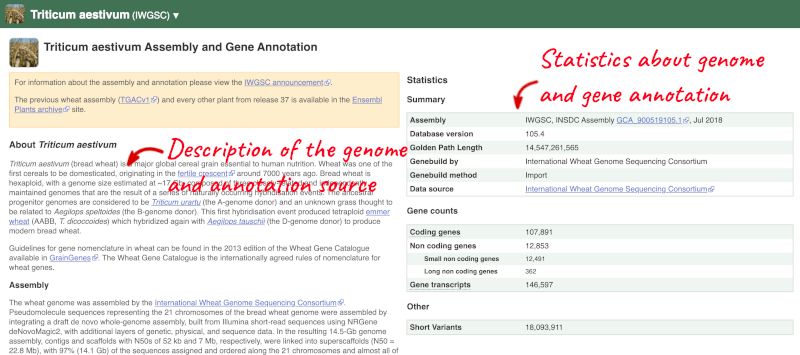
Here you’ll find a detailed description of how to the genome was produced and links to the original source. You will also see details of how the genes were annotated.
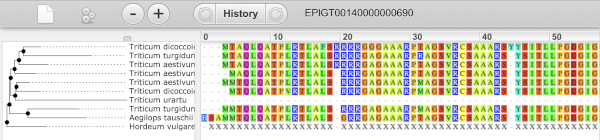
We’re going to look at the wheat TraesCS3D02G273600 gene. From plants.ensembl.org or the wheat species homepage, type _ TraesCS3D02G273600_ into the search bar and click the Go button.
The gene tab
Click on TraesCS3D02G273600 from the search hits. The Gene tab should open:
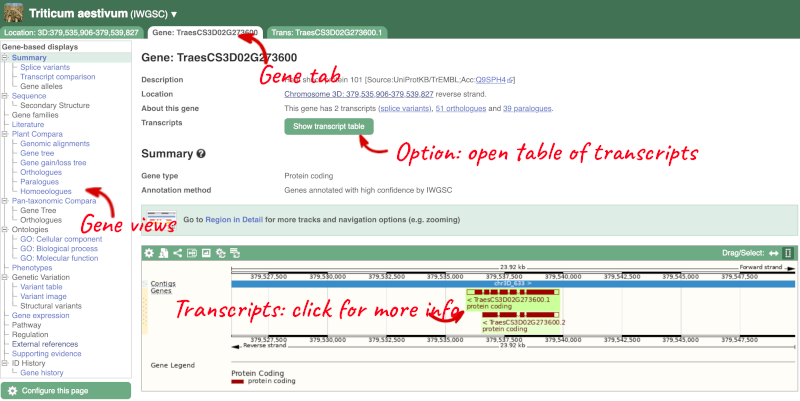
This page summarises the gene, including its location, name and equivalents in other databases. At the bottom of the page, a graphic shows a region view with the transcripts. We can see exons shown as blocks with introns as lines linking them together. Coding exons are filled, whereas non-coding exons are empty. We can also see the overlapping and neighbouring genes and other genomic features.
There are different tabs for different types of features, such as genes, transcripts or variants. These appear side-by-side across the blue bar, allowing you to jump back and forth between features of interest. Each tab has its own navigation column down the left hand side of the page, listing all the things you can see for this feature.
Let’s walk through this menu for the gene tab. How can we view the genomic sequence? Click Sequence at the left of the page.
The sequence is shown in FASTA format. The FASTA header contains the genome assembly, chromosome, coordinates and strand (1 or -1) – this gene is on the positive strand.
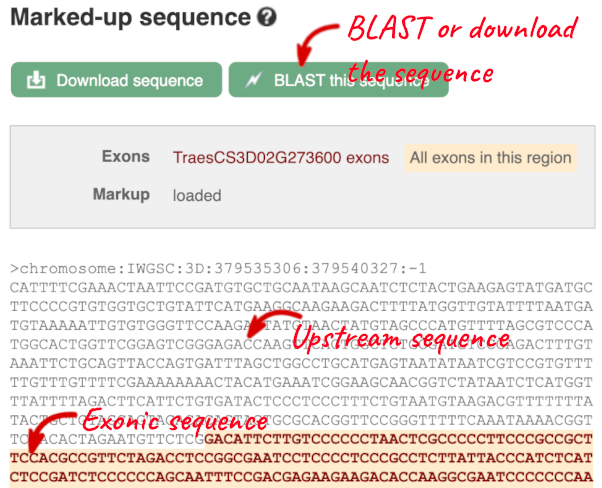
Exons are highlighted within the genomic sequence, both exons of our gene of interest and any neighbouring or overlapping gene. By default, 600 bases are shown up and downstream of the gene. We can make changes to how this sequence appears with the blue Configure this page button found at the left. This allows us to change the flanking regions, add variants, add line numbering and more. Click on it now.
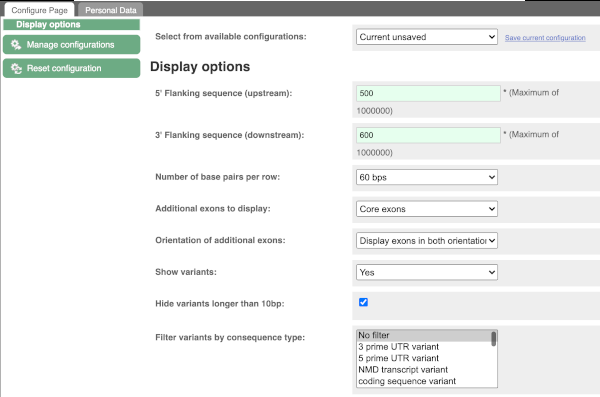
Once you have selected changes (in this example, Show variants and Line numbering) click at the top right.

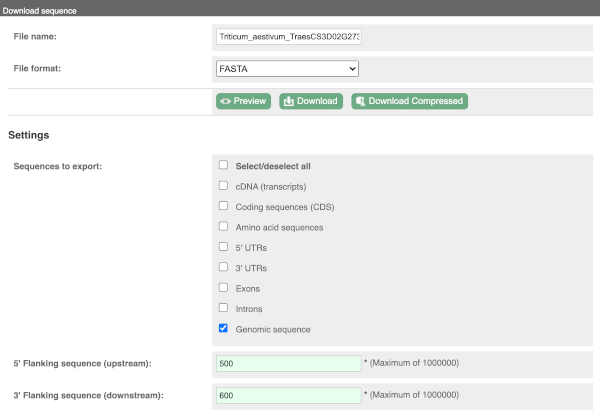
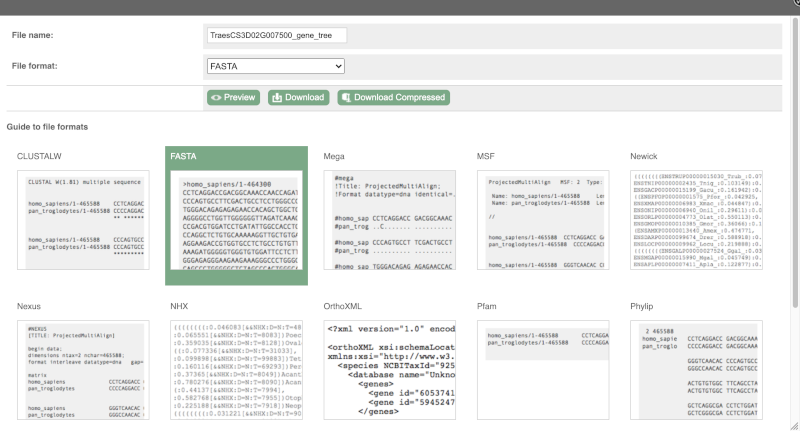
You can download this sequence by clicking in the Download sequence button above the sequence:

This will open a dialogue box that allows you to pick between plain FASTA sequence, or sequence in RTF, which includes all the coloured annotations and can be opened in a word processor. If you want run a sequence analysis tool, download as FASTA sequence, whereas if you want to analyse the sequence visually, RTF is best for this. This button is available for all sequence views.
To find out what the protein does, have a look at GO terms from the Gene Ontology consortium. There are three pages of GO terms, representing the three divisions in GO: Biological process (what the protein does), Cellular component (where the protein is) and Molecular function (how it does it). Click on GO: Biological process to see an example of the GO pages.
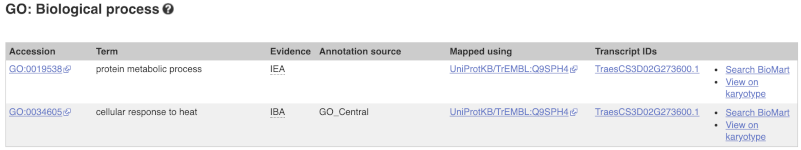
Here you can see the functions that have been associated with the gene. There are three-letter codes that indicate how the association was made, as well as links to the specific transcript they are linked to.
We also have links out to other databases which have information about our genes and may focus on other topics that we don’t cover, like Gene Expression Atlas or OMIM. Go up the left-hand menu to External references:
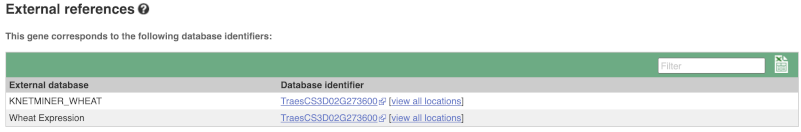
Demo: The transcript tab
We’re now going to explore the different transcripts of TraesCS3D02G273600. Click on Show transcript table at the top.
![]()
![]()
Here we can see a list of all the transcripts of TraesCS3D02G273600 with their identifiers, lengths and biotypes. Click on the ID of the largest transcript, HORVU5Hr1G100140.2.
You are now in the Transcript tab for TraesCS3D02G273600.1. We can still see the gene tab so we can easily jump back. The left hand navigation column provides several options for the transcript TraesCS3D02G273600.1 - many of these are similar to the options you see in the gene tab, but not all of them. If you can’t find the thing you’re looking for, often the solution is to switch tabs.
Click on the Exons link. This page is useful for designing RT-PCR primers because you can see the sequences of the different exons and their lengths.
![]()
You may want to change the display (for example, to show more flanking sequence, or to show full introns). In order to do so click on Configure this page and change the display options accordingly.
Now click on the cDNA link to see the spliced transcript sequence with the amino acid sequence. This page is useful for mapping between the RNA and protein sequences, particularly genetic variants.
![]()
UnTranslated Regions (UTRs) are highlighted in dark yellow, codons are highlighted in light yellow, and exon sequence is shown in black or blue letters to show exon divides. Sequence variants are represented by highlighted nucleotides and clickable IUPAC codes are above the sequence.
Next, follow the General identifiers link at the left. Just like the External References page in the gene tab, this page shows links out to other databases such as RefSeq, UniProtKB, PDBe and others, this time linked to the transcript or protein product, rather than the gene.
![]()
If you’re interested in protein domains, you could click on Protein summary to view domains from Pfam, PROSITE, Superfamily, InterPro, and more. These are all plotted against the transcript sequence, with the exons shown in alternating shades of purple at the top of the page. Alternatively, you can go to Domains & features to see a table of the same information.
![]()
Demo: The location tab
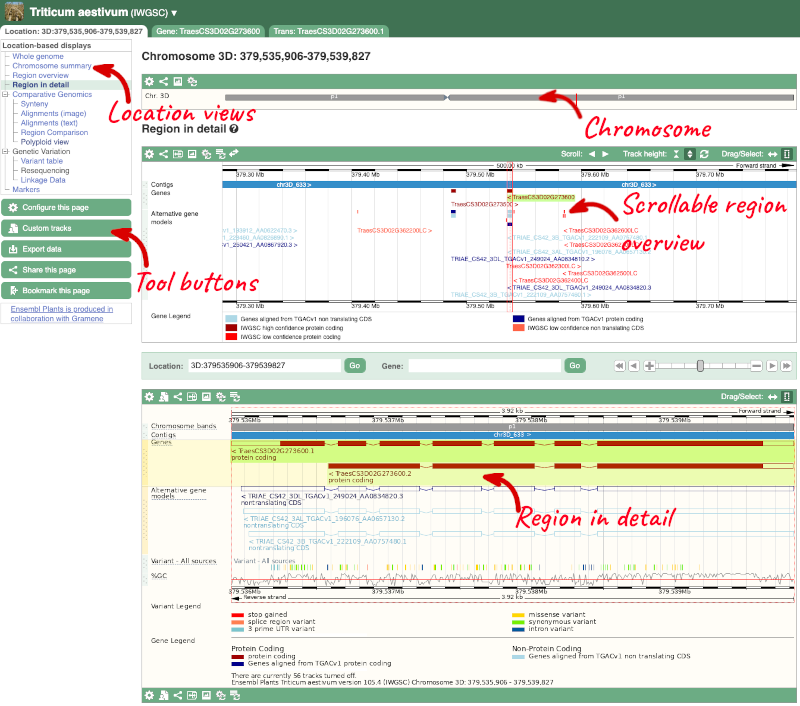
Click on the Location tab to view the Region in Detail page, which displays the genes and other related data aligned against the reference genome.
Click on the  button to view page-specific help. The help pages provide text, labelled images and, in some cases, help videos to describe what you can see on the page and how to interact with it.
button to view page-specific help. The help pages provide text, labelled images and, in some cases, help videos to describe what you can see on the page and how to interact with it.
The Region in detail page is made up of three images, let’s look at each one in detail.
1) The first image shows the chromosome:

The region we’re looking at is highlighted on the chromosome.
2) The second image shows a 500kb region around our selected region. This view allows you to scroll back and forth along the chromosome.
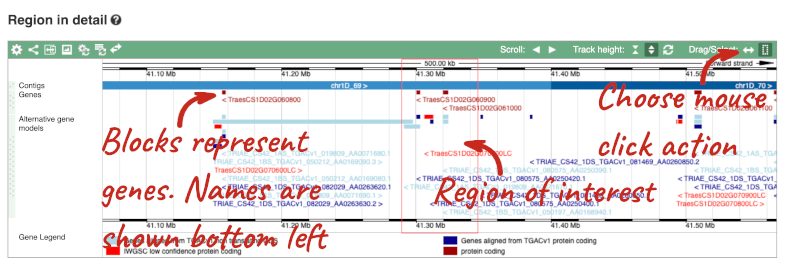
Click on the Drag/Select button  to change the action of your mouse click. Now you can scroll along the chromosome by clicking and dragging within the image. As you do this you’ll see the image below grey out and two blue buttons appear. Clicking on Update this image would jump the lower image to the region central to the scrollable image. We want to go back to where we started, so we’ll click on Reset scrollable image.
to change the action of your mouse click. Now you can scroll along the chromosome by clicking and dragging within the image. As you do this you’ll see the image below grey out and two blue buttons appear. Clicking on Update this image would jump the lower image to the region central to the scrollable image. We want to go back to where we started, so we’ll click on Reset scrollable image.

3) The third image is a detailed, configurable view of the region.
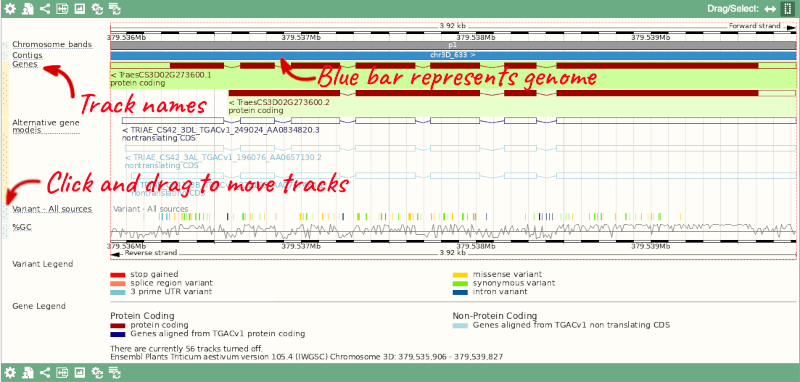
Here you can see various tracks, which is what we call a data type that you can plot against the genome. Some tracks, such as the transcripts, can be on the forward or reverse strand. Forward stranded features are shown above the blue contig track that runs across the middle of the image, with reverse stranded features below the contig. Other tracks, such as variants, regulatory features or conserved regions, refer to both strands of the genome, and these are shown by default at the very top or very bottom of the view.
You can use click and drag to either navigate around the region or highlight regions of interest, Click on the Drag/Select option at the top or bottom right to switch mouse action. On Drag, you can click and drag left or right to move along the genome, the page will reload when you drop the mouse button. On Select you can drag out a box to highlight or zoom in on a region of interest.
With the tool set to Select, drag out a box around an exon and choose Mark region.
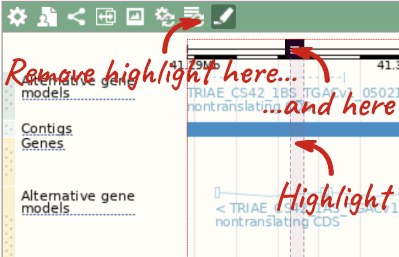
The highlight will remain in place if you zoom in and out or move around the region. This allows you to keep track of regions or features of interest.
We can edit what we see on this page by clicking on the blue Configure this page menu at the left.

This will open a menu that allows you to change the image.
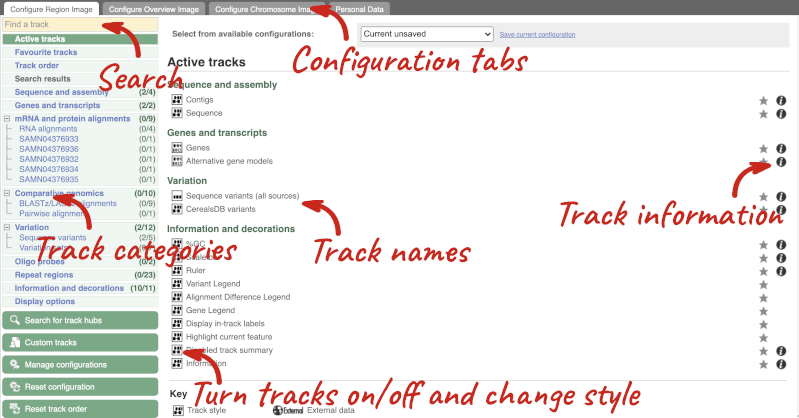
When you launch the view, you will see the tracks that are currently turned on with their names on the left and an info icon on the right, which you can click on to expand the description of the track. Turn them on or off, or change the track style by clicking on the box next to the name. More details about the different track styles are in this FAQ: http://www.ensembl.org/Help/Faq?id=335.
You can find more tracks to add by either exploring the categories on the left, or using the Find a track option at the top left. Type in a word or phrase to find tracks with it in the track name or description.
Let’s add some tracks to this image. Add:
- EMS-induced mutation variants
- Type I Transposons/LINE (Repeats: Repbase)
Now click on the tick in the top left hand to save and close the menu. Alternatively, click anywhere outside of the menu. We can now see the tracks in the image.
If the track is not giving you can information you need, you can easily change the way the tracks appear by hovering over the track name then the cog wheel to open a menu. To make it easier to compare information between tracks, such as spotting overlaps, you can move tracks around by clicking and dragging on the bar to the left of the track name.

Due to hybridisations in wheat’s evolutionary history, it has a hexaploid genome with related homoeologous regions. We can compare these with the Polyploid view. Click on the Polyploid view link in the left-hand menu.
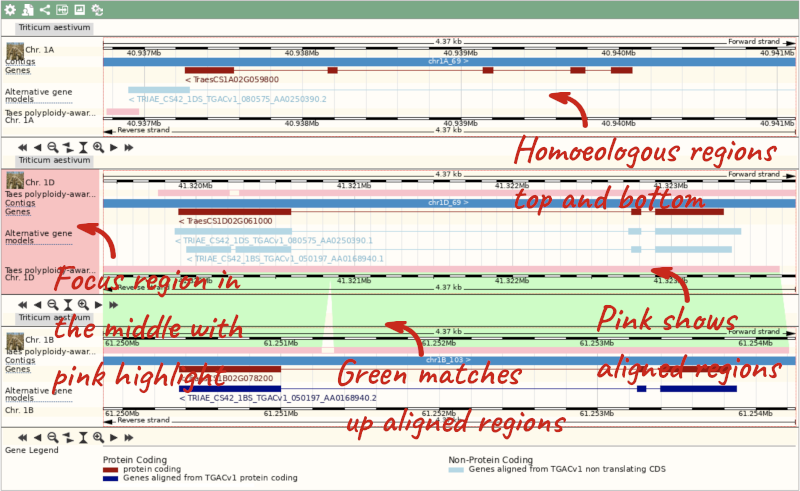
This view also allows us to configure the page, as we could with the main region view, so that we can compare other features between the homoeologous chromosomes.
Exploring the CCD7 gene in Arabidopsis thaliana
-
Find the Arabidopsis thaliana CCD7 gene on Ensembl Plants. On which chromosome and which strand of the genome is this gene located?
-
Where in the cell is the CCD7 protein located?
-
What is the source of the assigned gene name?
-
How many transcripts does it have? How long is its longest transcript (in bp)? How long is the protein it encodes? How many exons does it have? Are any of the exons completely or partially untranslated?
- Go to the Ensembl Plants homepage (http://plants.ensembl.org/). Select A. thaliana from the species list and type
CCD7in the search box. Click Go and click on the gene ID AT2G44990. You can find the strand orientation and the location under Summary in the Gene tab.The A. thaliana CCD7 gene is located on chromosome 2 on the forward strand.
- Click on GO: Cellular component in the left-hand panel.
The protein is located in the chloroplast and plastid.
- Click on Summary in the side menu.
The gene name is assigned and imported from NCBI gene (formerly Entrezgene).
- Click on Show transcript table.
There are 3 transcripts. The longest one is 2005 bp and the length of the encoded protein is 622 amino acids.
Click on the transcript ID AT2G44990.3 in the transcript table. You can find the number of exons in under in the summary information at the top of the page.
It has 6 exons.
Click on Sequence: Exons in the left-hand panel.
The first and last exons are partially untranslated (sequence shown in orange). This can also been seen from the fact that in the transcript diagrams on the Gene Summary and Transcript Summary pages the boxes representing the first and last exon are partially unfilled.
Exploring a plant gene (Vitis vinifera, grape)
Start in http://plants.ensembl.org/index.html and select the Vitis vinifera genome.
(a) What GO: biological process terms are associated with the MADS4 gene?
(b) Go to the transcript tab for the only transcript, Vv01s0010g03900.t01. How many exons does it have? Which one is the longest? How much of that is coding?
(c) What domains can be found in the protein product of this transcript? How many different domain prediction methods agree with each of these domains?
(a) Go to http://plants.ensembl.org/index.html.
Select Vitis vinifera from the drop down menu All genomes – select a species or click on View full list of all Ensembl Plants species and then choose V. vinifera.
Type MADS4 and click on the gene link VIT_01s0010g03900. Click on GO: Biological process in the side menu.
There are seven terms listed including GO:0006351, transcription, DNA-templated, and GO:0006355, regulation of transcription, DNA-templated.
(b) Click on the transcript named Vv01s0010g03900.t01 (or on the Transcript tab). Click on Exons in the left hand menu.
There are eight exons. Exon 8 is longest with 303 bp, of which 13 are coding.
(c) Click on either Protein Summary or Domains & features in the left hand menu to see graphically or as a table respectively.
A MADS-box domain near the N-terminus is identified by eight domain prediction methods. A K-box domain near the C-terminus is identified by two. Two coiled-coils are identified by one.
Finding a Triticum aestivum gene
-
Search for
Oxygen evolving enhancer proteinfrom the Ensembl Plants homepage and narrow down your search to Triticum aestivum. How many genes are there with this name in wheat? Why do you think this is? What chromosomes are they on? -
Go to the gene on chromosome 2B. How many protein-coding transcripts does this gene have? What is a “canonical transcript”?
-
Click on the canonical transcript. How many exons does this transcript have? Export the protein sequence of this transcript in the FASTA format.
- Start at the Ensembl Plants homepage. Choose Triticum aestivum from the species drop-down, type
Oxygen evolving enhancer proteininto the search box then click Go.There are two genes named TraesCS2D02G248400 and TraesCS2B02G270300. This is because of the hybridisations in wheat’s evolutionary history. You can see that the two genes occur on chromosomes 2B and 2D.
- Click on the gene on chromosome 2B to go to the Gene tab. If the transcript table is hidden, click on Show transcript table to see it.
There are 2 protein coding transcripts.
Mouse over the Ensembl Canonical flag in the transcripts table to find a description.
The Ensembl canonical transcript is a single transcript chosen for each gene in each species. It is the most highly conserved, most highly expressed, has the longest coding sequence and is represented in other key resources (e.g. NCBI, UniProt)
- Click on TraesCS2B02G270300.2 in the transcript table. You can find the number of exons in the summary description at the top of the Summary page, or you can count the number of boxes (boxes represent exons, lines represent introns) in the Summary diagram.
TraesCS2B02G270300.2 has 2 exons.
Go to Sequence: Protein In the left-hand panel.
Click on the green Download sequence button above the protein sequence. Select FASTA from the drop-down in the pop-up menu and download the sequence to your local machine.
Grape MADS4 region
Go to the Location view for MADS4 in Vitis vinifera.
(a) What is the closest gene to MADS4 in the grape genome? Find the location (base pair coordinates) of the closest gene, and the source of this annotation.
(b) Look at the EST alignments. Where do we see ESTs aligned?
(c) Look at regions of conserved gene order between grapevine and Arabidopsis thaliana by clicking Synteny at the left of the page. How many chromosomes in Arabidopsis show synteny to grapevine chromosome 1 in Ensembl?
(a) Go to http://plants.ensembl.org/index.html
Select Vitis vinifera from the drop down menu All genomes – select a species or click on View full list of all Ensembl Plants species and then choose V. vinifera.
Type MADS4 and hit Go, then click on the location link 1:21368964-21386383.
You should be in the Region in Detail page. Look at the middle image.
The closest gene is VIT_01s0010g03910. Click on it to find the location (Chromosome 1: 21,412,823-21,416,284) and that it is a novel transcript annotated by IGGP.
(b) Stay in the same view. Click on Configure this page, then select EST alignments on the left and choose EST (grape). Save and close the menu.
Most ESTs align to the exons of the gene.
(c) Click Synteny. The default species aligned is rice, choose Arabidopsis thaliana from the drop-down on the right.
There are 4 Arabidopsis chromosomes that show synteny to Grapevine chromosome 1. These are Arabidopsis chromosomes 1, 2, 3 and 5.
Exploring a wheat region
-
Go to 2D:378720500-378780600 in Triticum aestivum (wheat).
-
How many genes are in this region? What strand are the genes on? What are the gene IDs for these genes?
-
What tracks can you see that show gene structure? Where did the different tracks come from?
-
Export the genomic sequence for this region.
-
Can you view the genomic alignments of the homoeologous regions? What are the different formats you can export the image as?
-
Go to the Ensembl Plants homepage. Select Search: Triticum aestivum and type
2D:378720500-378780600in the text box. Click Go. -
There are two genes displayed in the Genes track. They are both located on the reverse strand. The IDs are
-
There are two tracks which have mapping to this gene: Genes and Alternative gene models. Click the track names for more information on their source.
-
Click Export data in the left-hand menu. Leave the default parameters as they are. Click Next>. Click on Text. Note that the sequence has a header that provides information about the genome assembly, the chromosome, the start and end coordinates and the strand. For example:
>2D dna:chromosome chromosome:IWGSC:2D:378720500:378780600:1 -
Click on Polyploid view in the left hand menu to view the homoeologous regions. Click on Export image. This will open a pop-up menu of the different image formats you can export, which are PNG and PDF.
Exploring a genomic region in Oryza sativa Japonica (rice)
Go to the Ensembl Plants homepage and do the following:
-
Go to the region between 405000 and 453000 on chromosome 1 in Oryza sativa Japonica.
-
Turn on the AGILENT:G2519F-015241 microarray track. Are there any oligo probes that map to this region?
-
Highlight the region around any reverse strand probes you can see. Do they map to any Ensembl transcripts?
-
Go to the Ensembl Plants homepage. Select Oryza sativa Japonica from the Species drop-down list and type
1:405000-453000. Click Go. - Click on Configure this page to open the menu. You can find the AGILENT:G2519F-015241 track under Oligo probes in the left-hand menu, or by using the Find a track box at the top right. Turn on the track as Normal then save and close the menu. As the AGILENT:G2519F-015241 track is stranded, it appears at the top and bottom of the view.
There are 5 probes mapped to this region on the positive strand and one probe on the reverse strand.
- Drag a box around the reverse strand probe then click on Mark region to highlight.
The highlighted region maps to two transcripts: Os01t0107900-02 and Os01t0107900-01
Variation
Exploring variants in rice
Visualising variants in the Sequence view
In any of the sequence views shown in the Gene and Transcript tabs, you can view variants on the sequence. You can do this by clicking on Configure this page from any of these views.
Let’s take a look at the Gene sequence view for OS01G0775500 in rice. Search for OS01G0775500 and go to the Sequence view.
If you can’t see variants marked on this view, click on Configure this page and select Show variants: Yes and show links.
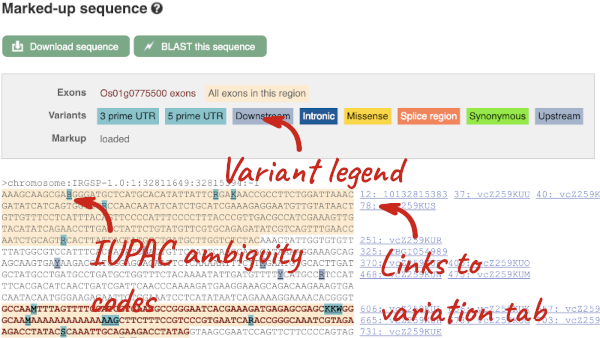
Find out more about a variant by clicking on it.
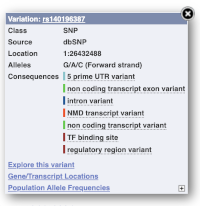
You can add variants to all other sequence views in the same way.
You can go to the Variation tab by clicking on the variant ID. For now, we’ll explore more ways of finding variants.
Viewing variants within a gene in the tabular form
To view all the sequence variants in table form, click the Variant table link at the left of the gene tab.
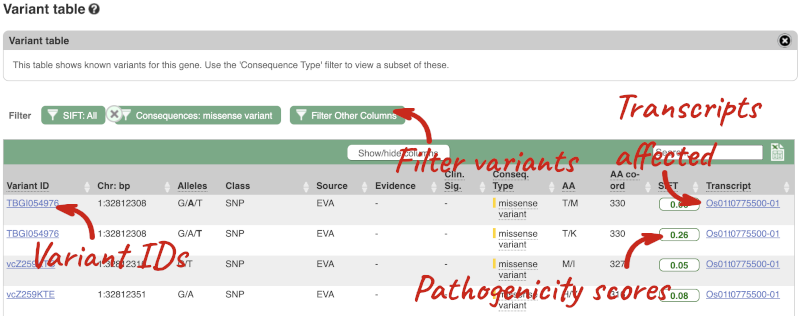
You can filter the table to only show the variants you’re interested in. For example, click on Consequences: All, then select the variant consequences you’re interested in. For display purposes, the table above has already been filtered to only show missense variants.
You can also filter by the different pathogenicity scores and MAF, or click on Filter other columns for filtering by other columns such as Evidence or Class.
The table contains lots of information about the variants. You can click on the IDs here to go to the Variation tab too.
Visualising variants in the Region in Detail view
Let’s have a look at variants in the Location tab. Click on the Location tab in the top bar.
Configure this page and open Variation from the left-hand menu.

There are various options for turning on variants. You can turn on variants by source, by frequency, presence of a phenotype or by individual genome they were isolated from. You can also turn on genotyping chips.
Exploring a specific variant
Let’s have a look at a specific variant. If we zoomed in we could see the variant rs18335701 in this region, however it’s easier to find if we put rs18335701 into the search box. Click through to open the Variation tab for Oryza sativa Japonica.
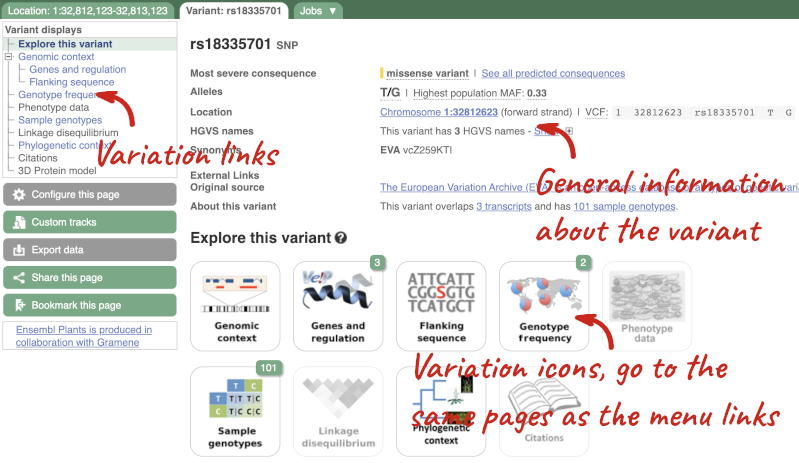
The icons show you what information is available for this variant. Click on Genes and regulation, or follow the link on the left.

This page illustrates the genes the variant falls within and the consequences on those genes, including pathogenicity predictors. It also shows data from GTEx on genes that have increased/decreased expression in individuals with this variant, in different tissues. Finally, regulatory features and motifs that the variant falls within are shown.
Let’s look at population genetics. Click on Population genetics in the left-hand menu.

The population allele frequencies are shown by study. Where genotype frequencies are available, these are shown in the tables.
We can see which strains these genotypes were observed in by going to Sample Genotypes. Click on Show for the Duitama et al. 2015 population.
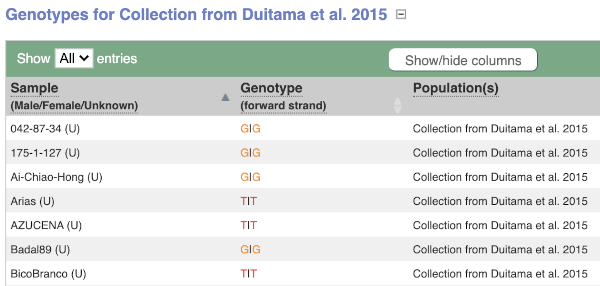
Exploring a SNP in Arabidopsis
The Arabidopsis thaliana ATCDSP32 protein is a chloroplastic drought-induced stress protein proposed to participate in a process called cell redox homeostasis. Go to Ensembl Plants and answer the following questions:
-
How many variants have been identified in the gene that can cause a change in the protein sequence (i.e. missense variant)?
-
What is the ID of the variant that changes the amino acid residue 60 from Alanine to Threonine (hint: refer to an amino acid codon table)? What is the location of this SNP in the A. thaliana genome? What are its possible alleles?
-
Download the flanking sequence of this SNP in RTF (Rich Text Format). Can you change how much flanking sequence is displayed on the browser?
-
Does this SNP cause a change at the amino acid level for other genes or transcripts?
- Click on Arabidospsis thaliana on the Ensembl Plants homepage. Search for ATCDSP32 on the species page and in the search results, click on the Gene ID AT1G76080. In the left-hand side menu of the Gene tab, click on Variant table. Click on Consequences: All then select only missense variant.
The missense variant button indicates that there are 18 of these. Alternatively, you can count the number of variants in your filtered list.
- An amino acid codon table can be found on Wikipedia. Sort the AA coord column by clicking on the header and scroll down to find a variant at residue 60. The ID of this variant is ENSVATH05153232.
The variant is located at position 28549171 on chromosome 1. The two possible alleles at this locus are C (reference) and T (alternative).
- Click on the link ENSVATH05153232, then click on Flanking sequence in the left-hand side menu. Now click on Download sequence and select File format > Rich Text Format (RTF).
If you want to change how much flanking sequence is displayed on the browser, go back to the Flanking sequence page, click on the Configuration button and change the length of the sequence. The default settings is 400 bp.
- Click on Genes and regulation in the left-hand side menu.
This SNP does not cause a change at the amino acid level for any other genes or transcripts in A. thaliana.
Variation data in tomato
-
Go to Ensembl Plants and find the Solyc02g084570.3 gene in Solanum lycopersicum (tomato) and go to its Location tab. Can you see the variation track?
-
Zoom in around the last exon of this gene. What are the different types of variants seen in that region? Are any splice region variants mapped in the region? If so, what is/are the coordinate(s)?
- Select Solanum lycopersicum from the Species search drop-down menu and search for
Solyc02g084570.3. In the results page, you can click on the coordinates 2:48284598-48288482 to go straight to the Location tab. Scroll down to the Region in detail view. The variation track is shown at the bottom of the view.If you don’t see the Variation - All sources track, click Configure this page on the left-hand panel, search for the track in the pop-up menu and enable the track by clicking on the square next to the track name. Close the pop-up window and wait for the track to load.
- Zoom in around the last exon of this gene by drawing a box in the respective region (you can change your mouse action by clicking the Drag/Select icons at the top right-hand corner of the view). Note the gene is on the reverse strand (this is signified by the < sign next to the transcript name, and it is located below the Contigs track), so the last exon will be on the left hand side of that image. The variation legend is shown at the bottom of the page, telling you what the colours mean.
The types of variants seen in that region are 3’ UTR, missense, synonymous and splice region variants.
Splice region variants are shown in orange. Click on the variants to get additional information on that variant including location. You can zoom into the region if the variant block is too small to click.
The variants are found at 2:48285642 and 2:48285640-48285641. Note that the two variants overlap: one is a SNP and the other is an indel. SNPs are tagged with ambiguity codes (zoom into the region if you cannot see this). You can find a useful IUPAC ambiguity code guid on the bioinformatics.org website. Single-letter ambiguity codes are given when two or more possible nucleotides may be represented at a single base locus.
Investigating a variant in wheat
-
Search for the variant
BA00369602in Triticum aestivum on Ensembl Plants. Is this variant known by any other names? -
What gene is affected by this variant? What is the amino acid change?
-
Which cultivars have the alternative base at this locus?
- Start at the homepage and enter BA00369602 into the search box and select Triticum aestivum from the drop-down list. Click on the Gene ID BA00369602 in the search results to get to the variation homepage.
Under Synonyms, you can see that the variant is also known as AX-94448191 in CerealsDB.
- Click on Genes and regulation.
The variant is a missense variant on TraesCS2D02G303800, where it gives a glycine to aspartic acid (G/D) change at transcript position 406.
- Click on Sample genotypes. Scroll down the table to see if there are any cultivars with the A allele in the genotype column.
All of the cultivars listed have the genotype
G|G.
VEP
Demonstration of the VEP web interface
Input
We have identified three variants on wheat chromosome 4B:
C -> T at 240206468
C -> G at 240199078
C -> T at 240212229
We will use the Ensembl VEP to determine:
- Have my variants already been annotated in Ensembl?
- What genes are affected by my variants?
- Do any of my variants affect gene regulation?
Click on Tools in the top green bar from any Ensembl Plants page, then Variant Effect Predictor to open the input form:
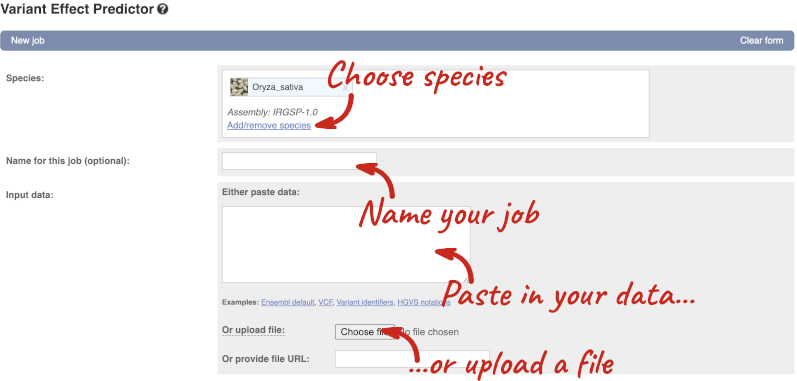
Click on Add/remove species and search for Triticum aestivum to choose it.
The data is in VCF:
chromosome coordinate id reference alternative
Put the following into the Paste data box:
4B 240206468 var1 C T
4B 240199078 var2 C G
4B 240212229 var3 C T
The VEP will automatically detect that the data is in VCF.
Additional configurations
There are further options that you can choose for your output. These are categorised as Identifiers, Variants and frequency data, Additional annotations, Predictions, Filtering options and Advanced options. Let’s open all the menus and take a look.
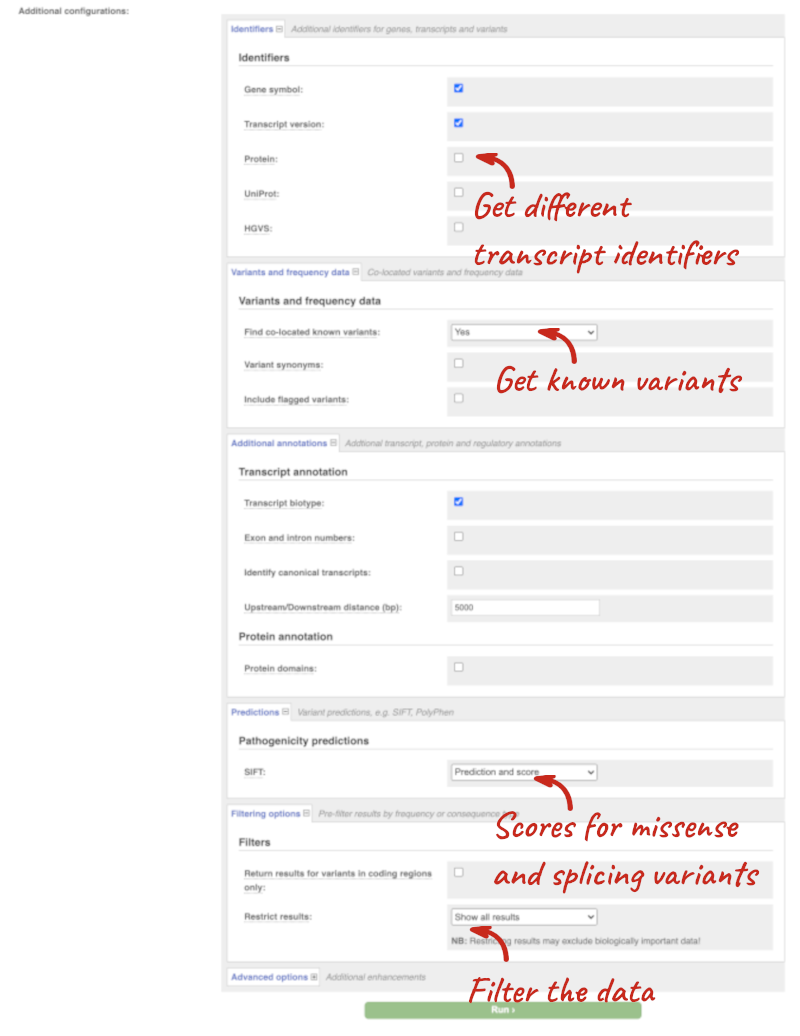
Hover over the options to see definitions.
When you’ve selected everything you need, scroll right to the bottom and click Run.

The display will show you the status of your job. It will say Queued, then automatically switch to Done when the job is done, you do not need to refresh the page. You can edit or discard your job at this time. If you have submitted multiple jobs, they will all appear here.
Click View results once your job is done.
Results
In your results you will see a graphical summary of your data, as well as a table of your results.
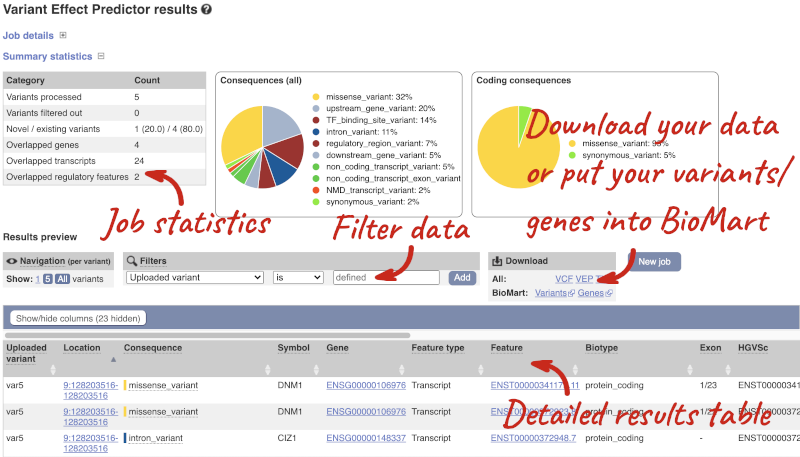
The results table is enormous and detailed, so we’re going to go through the it by section. The first column is Uploaded variant. If your input data contains IDs, like ours does, the ID is listed here. If your input data is only loci, this column will contain the locus and alleles of the variant. You’ll notice that the variants are not neccessarily in the order they were in in your input. You’ll also see that there are multiple lines in the table for each variant, with each line representing one transcript or other feature the variant affects.
You can mouse over any column name to get a definition of what is shown.
The next few columns give the information about the feature the variant affects, including the consequence. Where the feature is a transcript, you will see the gene symbol and stable ID and the transcript stable ID and biotype. The IDs are links to take you to the gene or transcript homepage.
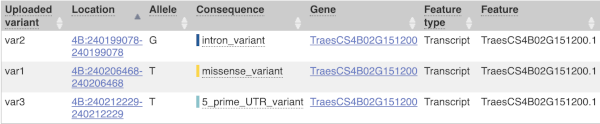
This is followed by details on the effects on transcripts, including the position of the variant in terms of the exon number, cDNA, CDS and protein, the amino acid and codon change and pathogenicity scores. Where the variant is known, the ID of the existing variant is listed, with a link out to the variant homepage. The pathogenicity scores are shown as numbers with coloured highlights to indicate the prediction, and you can mouse-over the scores to get the prediction in words.
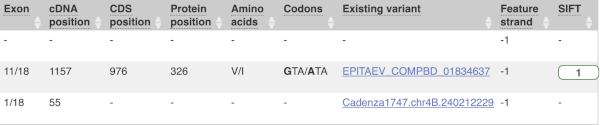
Above the table is the Filter option, which allows you to filter by any column in the table. You can select a column from the drop-down, then a logic option from the next drop-down, then type in your filter to the following box. We’ll try a filter of Consequence, followed by is then missense_variant, which will give us only variants that change the amino acid sequence of the protein. You’ll notice that as you type missense_variant, the VEP will make suggestions for an autocomplete.
You can export your VEP results in various formats, including VCF. When you export as VCF, you’ll get all the VEP annotation listed under CSQ in the INFO column. After filtering your data, you’ll see that you have the option to export only the filtered data. You can also drop all the genes you’ve found into the Gene BioMart, or all the known variants into the Variation BioMart to export further information about them.
Web VEP analysis of variants in Oryza sativa Japonica (rice)
You’ll find a VCF file here. This is a small subset of the outcome of Oryza sativa Japonica whole-genome sequencing and variant-calling experiment. Analyse the variants in this file with the VEP tool in Ensembl Plants and determine the following:
-
How many genes and transcripts are affected by variants in this file?
-
Do these variants result in a change in the proteins encoded by any of the Ensembl genes? Which genes are affected? What is the amino acid change? What is the pathogenicity prediction score for this change?
Go to Ensembl Plants and click on Tools at the top of the page. Click on Variant Effect Predictor and select Oryza sativa Japonica Group from the Species menu.
Either click on Choose file and select the file to upload it, or directly paste the URL into the Or provide file URL: box. Click Run at the bottom of the page. When your job is done, click View results.
- The number of affected genes and transcripts is shown in the Summary statistics table at the top.
8 genes and 8 transcripts are affected by these variants.
- Use the filters to view only missense variants. The filters are found above the detailed results table in the middle. Select Consequence and is from the drop-down menus. Then type
missense_variantinto the boxe. Add to apply your filter.1 variant is a missense variant. It causes a leucine to arginine (L/R) at position 16 change in the gene OS09G0103500. The SIFT score is 0.01 (Deleterious low confidence). Refere to this link for more information on SIFT (https://sift.bii.a-star.edu.sg/).
Web VEP analysis of variants in Triticum aestivum (wheat)
You have done whole-genome sequencing and variant-calling experiments for Triticum aestivum. You have a VCF file with a small subset of variants from this experiment. Analyse the variants in this file with the VEP tool in Ensembl Plants and determine the following:
-
How many variants were analysed? How many are novel?
-
How many genes and transcripts are affected by variants in this file?
-
Do any of the variants have different consequences for different transcripts?
-
Filter the table to find variants with high impact. How many variants have high impact? Why do you think missense variants are not classified as high impact?
-
Can you export all the results to a VCF file? Compare it to the input VCF file to see what information the VEP adds.
Go to any Ensembl Plants page and click on Tools in the navigation bar at the top of the page. Click on Variant Effect Predictor and change your species to Triticum aestivum by clicking on Change species.
Enter a descriptive name for your VEP job. If you have downloaded the variant file to your local machine, click on Choose file to upload. Alternatively, you can paste the URL for the file into the Or provide file URL: box. Click Run at the bottom of the page. When your job is done, click View reesults.
-
20 variants were analysed, of which 1 is novel.
-
Only 1 gene is affected by variants in this file. The gene has 2 transcripts and both are affected by the variants.
- You can find a list of calculated variant consequences and their impact here.
Yes, the novel variant results in a stop_lost in TraesCS3A02G301400.1 and is a downstream_gene_variant for TraesCS3A02G301400.2.
- Use the filters to view only variants with HIGH impact (you may need to add the column under Show/hide columns at the top of the table if you cannot find it). The filters are found above the detailed results table in the middle. Select Impact and is from the drop-down menus. Then type
HIGHinto the box; this will autocomplete. Click Add.There are 3 variants with high impact and all three are stop altering. Missense variants are not classified as high impact, because they do not always have significant impacts on protein functions. Usually the protein is still produced. In contrast, stop altering variants affect the protein length, and therefore likely affect the protein function.
- At the top right of the table there is an option to download data. Click on VCF for the All option. Open the VCF file you have downloaded in a text editor. You can see that VEP adds annotation in the INFO column of the VCF file.
Plants comparative genomics
Demo: gene trees and homology predictions
Plants Compara
Gene trees
Let’s look at the homologues of Triticum aestivum (wheat) TraesCS3D02G007500. Open Ensembl Plants, search for the gene and go to the Gene tab.
Click on Plant compara: Gene tree, which will display the current gene in the context of a phylogenetic tree used to determine orthologues, paralogues and homoeologues.

Funnels indicate collapsed nodes. We can expand them by clicking on the node and selecting Expand this sub-tree from the pop-up menu.
We can also see the protein alignment of the sub-tree by clicking on Wasabi viewer, which will open a pop-up:
You can download the tree in a variety of formats. Click on the download icon ![]() in the bar at the top of the image to get a pop-up where you can choose your format.
in the bar at the top of the image to get a pop-up where you can choose your format.
Homologues
We can look at homologues in the Orthologues, Paralogues and homoeologues pages, which can be accessed from the left-hand menu. If there are no orthologues, paralogues or homoeologues, then the name will be greyed out. Click on Plant compara: Orthologues to see the orthologues available in plants.
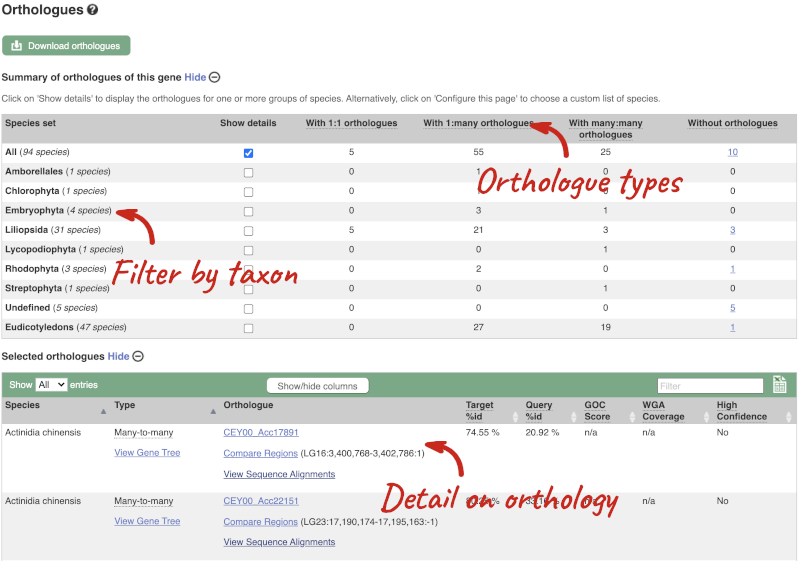
Choose to see only Eudicotyledons orthologues by selecting the box. The table below will now only show details of Eudicotyledons orthologues. Let’s look at Brassica oleracea.
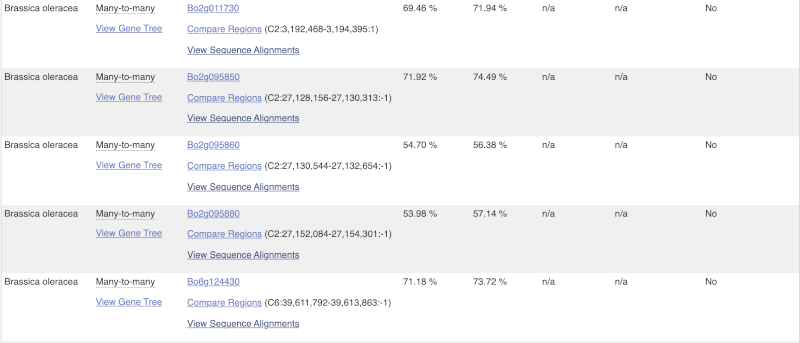
Here we can see there is a many-to-many relationship between the wheat and B. oleracea orthologues. Links from the orthologue allow you to go to alignments of the orthologous proteins and cDNAs. Click on View Sequence Alignments then View Protein Alignment for the first B. oleracea orthologue.
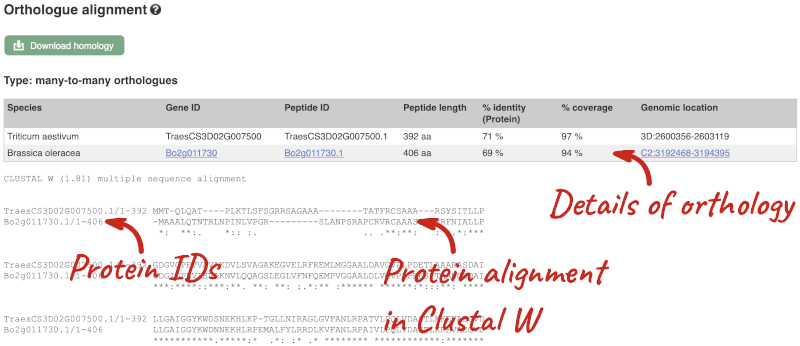
The paralogue page and homoeologue pages are structured in the same way as the orthologue page.
Let’s look at some of the comparative genomics views in the Location tab. Go to the region 1:8000-18000 in Oryza sativa Japonica.
We can look at individual species comparative genomics tracks in this view by clicking on Configure this page.
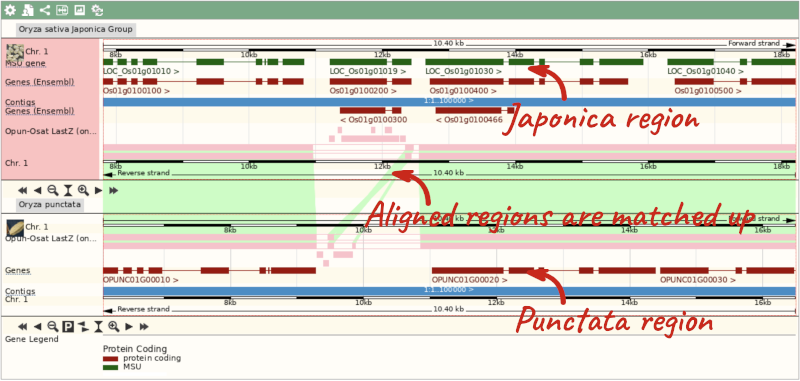
Select BLASTz/LASTz alignments from the left-hand menu to choose alignments between closely related species. Turn on the alignments for all Oryza species:

The alignment is greatest between closely related species. We can see that many rice species (such as Oryza barthii) are fully aligned across the region, but other species have a region around 1:10700-12500 where a different chunk is aligned (such as Oryza punctata).
We can also look at the alignment between species or groups of species as text. Click on Alignments (text) in the left hand menu.
Select Select an alignment to open the alignment menu.
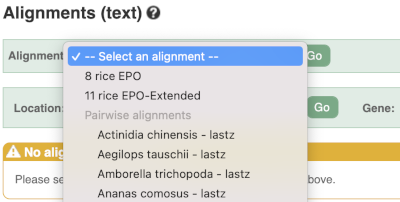
Select Oryza punctata from the alignments list then click Go.
In this case there are eight blocks aligned of different lengths, some of which correspond to the region we saw unaligned in the image. Click on Block 1.
You will see a list of the regions aligned, followed by the sequence alignment. Click on Display full alignment. Exons are shown in red.
To compare with both contigs visually, go to Region comparison.
To add species to this view, click on the blue Select species or regions button. Choose Oryza punctata again then close the menu.
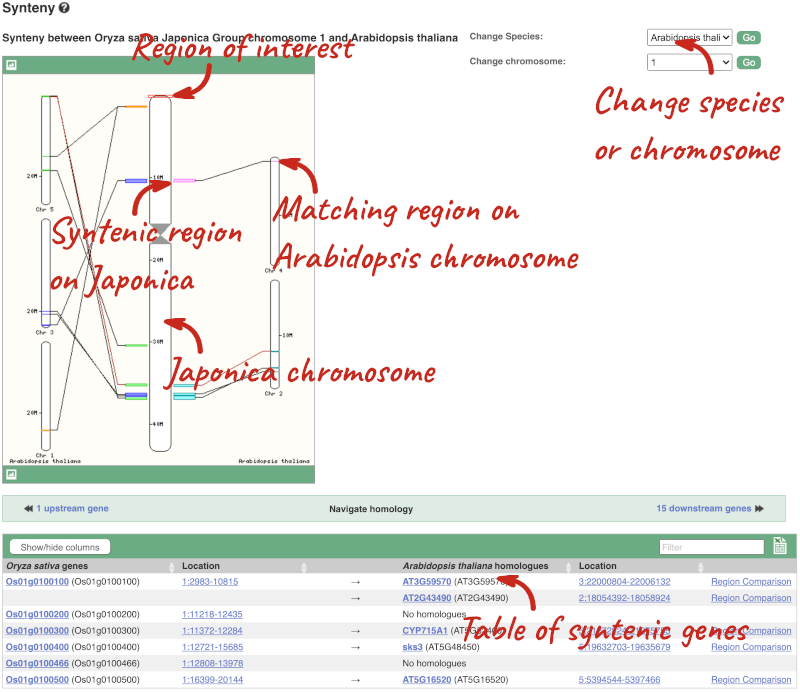
We can view large scale syntenic regions from our chromosome of interest. Click on Synteny in the left hand menu.
Finding orthologues and gene trees of the Arabidopsis thaliana FUM1 gene
The fumarase gene FUM1 in Arabidopsis thaliana encodes a protein with mitochondrial targeting information. Read more in this UniProt entry. Go to Ensembl Plants to answer the following questions:
-
How many orthologues have been identified for this gene?
-
Which orthologue has the highest sequence similarity? Look at the Query%ID and Target%ID.
- Go to Ensembl Plants, select Arabidopsis thaliana from the Favourite genomes section on the homepage. Search for
FUM1. Click on the gene ID AT2G47510. Now click on Plant Compara: Orthologues on the left-hand panel to see all orthologues of this gene. You can find the number of orthologues in the summary information at the top of the page.FUM1 has 166 orthologues in Ensembl Plants.
- Click on the triangles in the table column headers to sort by identity. If you are unsure of what data the column is show, you can mouse-over the headers for a description.
The orthologue with the highest sequence similarity is from Arabidopsis halleri.
Homologues and gene trees for the Triticum aestivum (wheat) RHT1 gene
Go to Ensembl Plants and answer the following questions:
-
How many orthologues are predicted for the Triticum aestivum (wheat) gene RHT1 (gene ID TraesCS4D02G040400) gene in Liliopsida?
-
How much sequence identity does the Secale cereale (rye) protein have to the maize one?
-
Download the alignment in Nexus format.
-
Open the gene tree for the wheat RHT1 gene. What is the gene tree ID?
-
How many speciation and duplication nodes does the phylogeny have?
Go to the Ensembl Plants homepage, select Triticum aestivum from the Species drop-down and search for TraesCS4D02G040400. Click through to the Gene tab. On the Gene tab, click on Plant Compara: Orthologues at the left-hand side of the page to see all the orthologous genes.
- These are the orthologues in the Liliopsida:
- 24 1-to-1
- 9 1-to-many
- 0 many-to-many
- Filter the table by entering
Secale cerealein the filter box on the top right-hand corner of the table.The percentage of identical amino acids in the rye protein (the orthologue) compared with the gene of interest (i.e. wheat RHT1; the target species/gene) is 98.71%. This is known as the Target %ID. The identity of the gene of interest (wheat RHT1) when compared with the orthologue (the rye gene, i.e. the query species/gene) is 97.91% (the query %ID).
Note the differences in the values of the Target and Query % ID reflects the different protein lengths for the genes. -
Click on View Sequence Alignments in the Orthologue column. Select View Protein Alignment from the pop-up menu. Click on the green Download homology button above the table and select Nexus. Click on Download or Download Compressed to save the alignment on your local machine.
- Go to Plant Compara: Gene tree in the left-hand menu. You can find the gene tree ID above the phylogeny.
The gene tree ID is EPlGT00940000163877.
- You can find some summary statistics below the gene ID.
There are 418 speciation nodes and 149 duplication nodes.
Exploring whole-genome alignments for Triticum aestivum (wheat)
Go to Ensembl Plants and answer the following questions:
-
Find the TraesCS2D02G080000 gene in Triticum aestivum (wheat). What is the function for this gene and what are its coordinates?
-
Go to the Location tab. Turn on the LASTZ-net alignment tracks for Arabidopsis thaliana, Zea mays (corn) and Sorghum bicolor (great millet). Are there any regions where you can see gaps in in some of the species alignments?
-
Go to the Region comparison view and compare to A. thaliana. What occurs at this gap in the alignment?
-
Export the Block 2 alignment between T. aestivum and A. thaliana in ClustalW format.
- Go to the Ensembl Plants homepage. Select Triticum aestivum from the Species drop-down, enter
TraesCS2D02G080000in the search box and click Go. Open the Gene tab.The gene description is as follows: Ascorbate peroxidase, ROS homeostasis, Chloroplast protection, Carbohydrate metabolism, Plant architecture, Fertility maintenance. This was projected from Oryza sativa (Os07g0694700).
- Go the Location tab in the top left-hand corner. Click on CConfigure this page in the side menu. Open Comparative genomics: BLASTz/LASTz alignments in the pop-up menu. Turn on the tracks for Arabidopsis thaliana, Zea mays (corn) and Sorghum bicolor (great millet) in the Normal style. Save and close the pop-up menu
There is alignment across most of the coding regions, with some gaps occurring in all 3 species. These gaps map with the intronic regions of the T. aestivum gene.
- Click on Comparative Genomics: Region Comparison in the left-hand menu. Go to the Select species or regions button and add A. thaliana. Save and close the menu.
The gap in the alignment translates to the intronic regions of the T. aestivum gene.
- Go to Comparative Genomics: Alignments (text) and select A. thaliana from the Alignment drop-down. Click on the green Download alignment button and select ClustalW. Download the file to your local machine either in a compressed format, or as it is by clicking the green Download button above the file format preview.
Orthologues, paralogues and gene trees for the maize Zm00001d015746 gene
How many orthologues are predicted for the maize Zm00001d015746 gene in Liliopsida?
How much sequence identity does the Sorghum bicolor protein have to the maize one? Click on the Alignment link next to the Ensembl identifier column to view a protein alignment in Clustal format.
Go to plants.ensembl.org, choose Zea mays and search for Zm00001d015746. Click through to the Gene tab view.
On the gene tab, click on Orthologues at the left side of the page to see all the orthologous genes.
These are the orthologues in the Liliopsida:
- 20 1-to-1
- Seven 1-to-many
- Two many-to-many
The percentage of identical amino acids in the sorghum protein (the orthologue) compared with the gene of interest. i.e. maize Zm00001d015746 (the target species/gene) is 82.07%. This is known as the Target %ID. The identity of the gene of interest (maize GRMZM2G144081) when compared with the orthologue (the sorghum gene, the query species/gene) is 81.69% (the query %ID).
Note the differences in the values of the Target and Query % ID reflects the different protein lengths for the genes.
Finding orthologous genes for disease resistance gene in Coffea canephora (coffee)
Resistance to the leaf rust delivered by SH3 factor(s) is well-grounded as specially durable. in 2023, Paula Cristina da Silva Angelo et al (https://doi.org/10.1016/j.pmpp.2023.102111) reported that the Arabidopsis thaliana gene AT1G50180 is an important gene in the SH3 locus conferring diseae resistance.
Search Ensembl Plants for the gene AT1G50180 in Arabidopsis thaliana.
-
From the gene tab, go to the Arabidopsis thaliana AT1G50180 gene Orthologues page under Plant Compara.
-
Reduce the orthologues table to look only at Coffea canephora (coffee) orthologues. How many results can you see?
-
Download the cDNA alignment in ClustalW format for the alignment between the Arabidopsis thaliana AT1G50180 gene and the Coffea canephora GSCOC_T00030728001 gene.
Go to Ensembl Plants. Select Arabidopsis thaliana from the drop-down box and type in AT1G50180. Click Go and click on the gene ID AT1G50180.
-
Go to Plant Compara: Orthologues on the left-hand panel.
- Filter for Coffea canephora using the filter option in the top right hand corner of the table.
Coffee has 25 many-to-many orthologues.
- Click on View Sequence Alignments then cDNA (found in the 3rd column below the gene identifier) for the GSCOC_T00030728001 gene. This takes us to the Orthologue Alignment page.
Click on Download Homology to download the alignment in ClustalW format
Finding orthologous genes for a root transporter in Oryza sativa Japonica (rice)
Search Ensembl Plants for the gene Lsi1 in Oryza sativa Japonica Group (rice). This gene is known to code for an aquaporin transporter that facilitates the uptake of silicon and arsenic through the roots. Silicon concentration is highest in grass species, and is associated with defence.
-
From the gene tab, go to the Orthologues page under Plant Compara. Which plant group has the highest number of 1-to-1 orthologues? Is it the same group that has the highest number of 1-to-many orthologues?
-
Reduce the orthologues table to look only at Triticum aestivum (wheat) orthologues. Why are there three results for a 1-to-1 orthologue?
-
Click on the Compare regions link for chromosome 6B region in wheat to go to the Location tab. Scroll to the bottom image. How do the gene models compare between the species? Do they have the same number of exons?
-
Click back to the Gene tab and click on the Gene gain/loss tree page. Which species has the highest number of members of this gene family? Is it a grass? Can you change the view to see a radial tree?
Go to Ensembl Plants. Look for the main search box highlighted in green. Select Oryza sativa Japonica Group from the drop-down box and type in Lsi1. Click Go and click on the gene ID Os02g0745100.
- Go to Plant Compara: Orthologues on the left-hand panel.
Liliopsida has 24 1-to-1 orthologues, the only group with 1-to-1 orthologues. This group is synonymous with Monocotyledon, so the group that contains the grasses. Eudicotyledons has the highest number of 1-to-many orthologues, indicating that this gene has been duplicated in the eudicots.
- Use the search box in the top right-hand corner of the Selected orthologues table and enter
Triticum aestivum, the table should automatically filter.There are 3 results, one for each component (A,B,D). Note that these are considered 1-to-1 orthologues, rather than 1-to-many. This is because these genes arose in wheat by hybridisation (allopolyploidy), rather than duplication (autopolyploidy).
- Click on Compare regions (found in the 3rd column below the gene identifier) from the 2nd result for component 6B. This takes us to the Location tab. Scroll down to the bottom of the page.
Both genes have 5 exons and the same structure. This looks unusual because the gene in rice is on the forward strand, while the gene in wheat is on the reverse strand. This is reflected in the crossing green links between the pink alignment blocks.
- Click on the Gene tab at the top of the page and click on Gene gain/loss tree in the left-hand panel.
Significant expansions are shown with red branches, and the number of genes in the family shown in the count next to the image and species name. We can see that Echinochloa crus-galli (Cockspur grass) has 25 members in this group.
We can change the tree to radial view by clicking on the icon with two arrows at the top left of the image.