Filter Events by Year
Ensembl Genome Browser Workshop – Graphic Era Hill University (GEHU)
Course Details
- Lead Trainer
- Jorge Batista da Rocha
- Associate Trainer(s)
- Event Dates
- 2024-11-25 until 2024-11-26
- Location
- Dehradun, India
- Description
- Work with the Ensembl Outreach team to get to grips with the Ensembl browser, accessing gene, regulation and comparative genomics data.
- Survey
- Ensembl Genome Browser Workshop – Graphic Era Hill University (GEHU) Feedback Survey
Demos and exercises
Ensembl species
Demo: Exploring species and genome assemblies in Ensembl Plants
Homepage
The front page of Ensembl Plants is found at plants.ensembl.org. It contains lots of information and links to help you navigate Ensembl Plants:
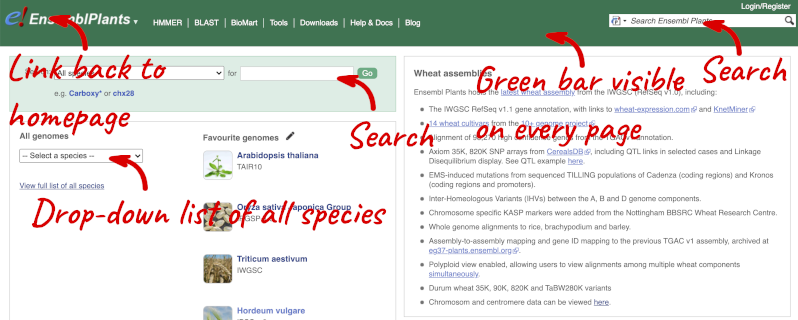
At the top left you can see the current release number and what has come out in this release.
Available species
Click on View full list of all species.
Click on the scientific name of your species of interest to go to the species homepage. We’ll click on Triticum aestivum.
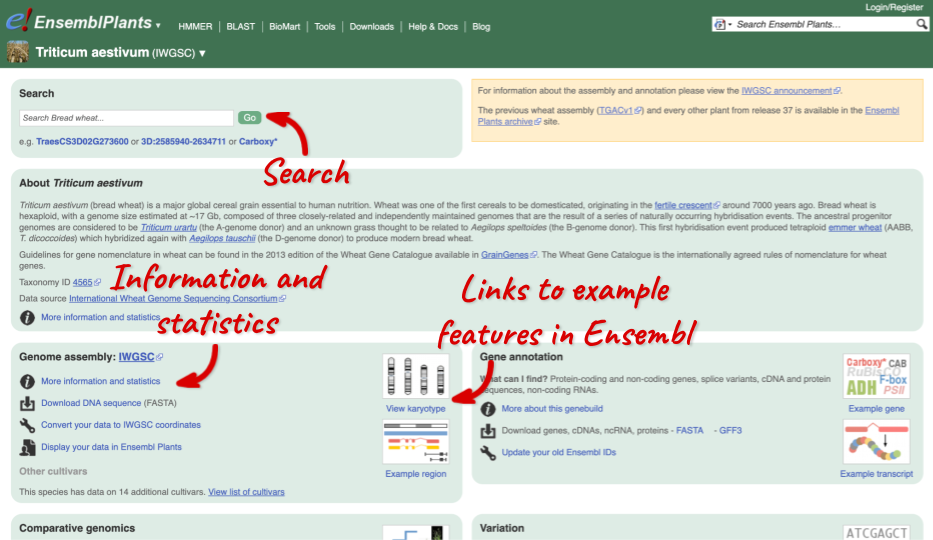
Species information
Here you can see links to example pages and to download flatfiles. To find out more about the genome assembly and genebuild, click on More information and statistics.
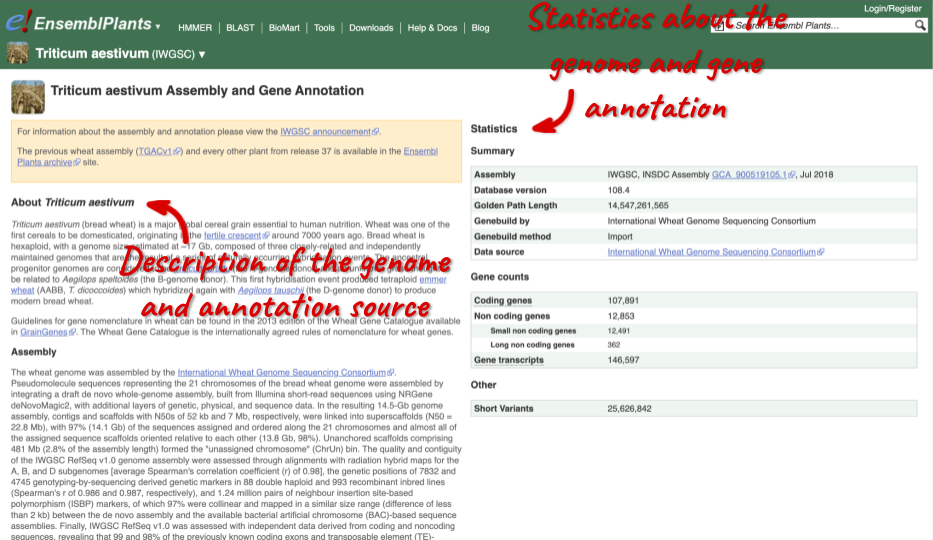
Here you’ll find a detailed description of how to the genome was produced and links to the original source. You will also see details of how the genes were annotated.
Oryza sativa Japonic (rice) gene counts
Find the species Oryza sativa Japonica in Ensembl Plants. How many coding and non-coding genes does it have?
Select Oryza sativa Japonica from the homepage to go to its species information page. Click on More information and statistics.
Oryza sativa Japonica has 37,960 coding and 1,011 non-coding genes.
Triticum aestivum (wheat) cultivars
-
Are there any additional cultivars available alongside the Triticum aestivum (IWGSC) reference genome?
-
Find the description of the wheat assembly. Which institute provided the assembly and annotations?
-
How many coding and non-coding genes does the IWGSC assembly have?
-
Are there any other species of the genus Triticum available in Ensembl? If so, which species are they?
- Go to Ensembl Plants and click on Triticum aestivum on the front page of Ensembl Plants to go to the species information page. Under the Genome assembly section of the species page, you will find the number of cultivars in wheat.
There are 14 cultivars.
- Click on More information and statistics in the Genome assembly section and scroll down to the paragraph on Assembly.
The assembly and annotations were generated by the International Wheat Genome Sequencing Consortium (IWGSC).
- Stay on the More information and statistics page. You can find some summary statistics on the right-hand side.
The T. aestivum (IWGSC) assembly has 107,891 coding and 12,853 non-coding genes.
- Go to the Ensembl Plants homepage. Click on View full list of all species in the All genomes panel. Filter the table by entering
Triticumin the text box on the top right-hand corner of the table.Besides T. aestivum are 4 other Triticum species available in Ensembl: Triticum dicoccoides (wild emmer wheat), Triticum spelta (spelt), Triticum turgidum (domesticated emmer wheat) and Triticum urartu (red wild einkorn wheat).
Solanum genus
Go to Ensembl Plants and answer the following questions:
-
How many genomes of the genus Solanum are there in Ensembl Plants?
-
When was the current Solanum lycopersicum genome assembly last revised?
- On the homepage, click on View full list of all Ensembl Plants species underneath the coloured search block. Type Solanum into the filter box in the top left-hand corner of the table.
There are three Solanum genomes: Solanum lycopersicum (tomato), and Solanum tuberosum RH89-039-16 and Solanum tuberosum (both potato).
- Click on S. lycopersicum, then on More information and statistics.
The genome was revised in April 2018.
Exploring the Coffee genome assembly
-
What is the name of the coffee variety represented in Ensembl?
-
Who produced this genome assembly and annotation?
-
What is the length of the Coffea canephora genome assembly? How many coding genes are annotated across the genome?
Select Coffea canephora from the drop down species list, or click on View full list of all species, then choose Coffea canephora from the list to go to the species homepage.
- The coffee variety represented in Ensembl Plants is Coffea canephora (Robusta coffee). The Arabica coffee variety is not currently represented in Ensembl Plants.
Click on on More information and statistics.
-
The AUK_PRJEB4211v1 _Coffea canephora assembly was submitted by Genoscope CEA.
-
The genome is 568,611,505bp in length. There are 25,574 coding genes annotated across the genome.
Finding a genome in Ensembl Bacteria
Mycobacterium tuberculosis H37Ra str. ATCC25177 is a clinical strain.
Go to Ensembl Bacteria and find the species M. tuberculosis H37Ra str. ATCC25177. How many coding genes does it have?
In the Ensesmbl Bacteria homepage, start to type H37Ra into the Search for a genome search box (you can find this in the coloured block at the top of the homepage). It will auto-complete, allowing you to select M. tuberculosis H37Ra str. ATCC25177 from the drop-down list. Click on More information and statistics.
M. tuberculosis H37Ra str. ATCC25177 has 4,080 coding and 47 non-coding genes.
Exploring the Botrytis cinerea genome
Botrytis cinerea is the causal agent of the grey mold disease and warty berry in coffee.
-
Who produced this genome assembly and annotation?
-
What is the length of the Botrytis cinerea genome assembly? How many coding genes are annotated across the genome?
Go to Ensembl Fungi [https://fungi.ensembl.org/index.html]. Select Botrytis cinerea from the drop down species list, or click on View full list of all species, then choose Botrytis cinerea from the list to go to the species homepage.
Click on on More information and statistics.
-
The ASM83294v1 Botrytis cinerea assembly was submitted by Wageningen University and Syngenta.
-
The genome is 42,630,066 bp in length. There are 11,707 coding genes annotated across the genome.
Chicken assembly
When was the current Gallus gallus genome assembly submitted and by whom?
Select Chicken from the drop down species list, or click on View full list of all Ensembl species, then choose Chicken from the list to go to the species homepage. Click on on More information and statistics.
The bGalGal1.mat.broiler.GRCg7b assembly was submitted by Vertebrate Genomes Project on January 2021.
Pig species data
-
How many coding and non-coding genes does pig have?
-
When was the current Sus scrofa genome assembly produced and by whom?
1.Select Pig from the drop down species list, or click on View full list of all Ensembl species, then choose Pig from the list to go to the species homepage. Click on More information and statistics.
Pig has 22,063 coding genes and 13,154 non-coding genes.
- The Sscrofa11.1 assembly of the pig genome was produced in January 2017 by the Swine Genome Sequencing Consortium (SGSC).
Sheep species data
(a) Go to the species homepage for Sheep. What is the name of the genome assembly for Sheep?
(b) Click on More information and statistics. How long is the Sheep genome (in bp)? How many genes have been annotated?
(a) Select Sheep from the drop down species list, or click on View full list of all Ensembl species, then choose Sheep from the list.
The assembly is Oar_rambouillet_v1.0 or GCA_002742125.1.
(b) Click on More information and statistics. Statistics are shown in the tables on the left.
The length of the genome is 2,869,914,396 bp. There are 20,506 coding genes.
Cod assembly
Go to the species homepage for Atlantic Cod. Click on More information and statistics. How long is the Cod genome (in bp)? How many genes have been annotated?
Select Atlantic cod from the drop down species list, or click on View full list of all Ensembl species, then choose Atlantic cod from the list to go to the species homepage. Click on on More information and statistics.
The length of the genome is 669,949,713 bp. There are 23,515 coding and 5,339 non-coding genes annotated.
Exploring genomic regions
Demo: Exploring genomic regions in Ensembl Plants
Start at the Ensembl Plants front page. You can search for a region by typing it into a search box, but you have to specify the species.
To bypass the text search, you need to input your region coordinates in the correct format, which is chromosome, colon, start coordinate, dash, end coordinate, with no spaces for example: 1D:41289600-41345600. Choose Triticum aestivum from the species drop-down, then type (or copy and paste) these coordinates into the search box.
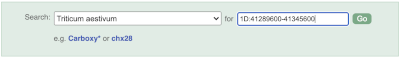
Press Enter or click Go to jump directly to the Region in detail Page.
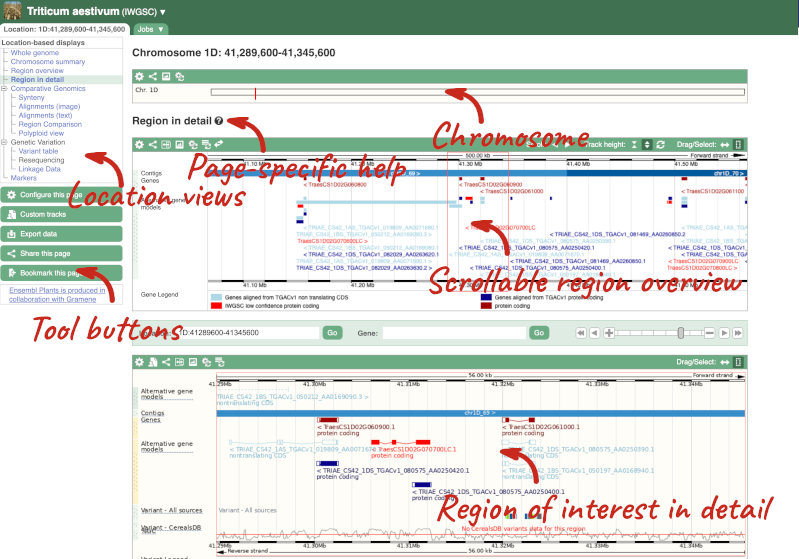
Click on the  button to view page-specific help. The help pages provide text, labelled images and, in some cases, help videos to describe what you can see on the page and how to interact with it.
button to view page-specific help. The help pages provide text, labelled images and, in some cases, help videos to describe what you can see on the page and how to interact with it.
The Region in detail page is made up of three images, let’s look at each one in detail.
- The first image shows the chromosome:

The region we’re looking at is highlighted on the chromosome. You can jump to a different region by dragging out a box in this image. Drag out a box on the chromosome, a pop-up menu will appear.
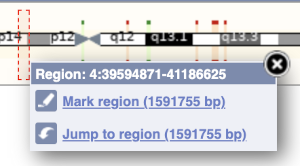
If you wanted to move to the region, you could click on Jump to region (### bp). If you wanted to highlight it, click on Mark region (###bp). For now, we’ll close the pop-up by clicking on the X in the corner.
- The second image shows a 1Mb region around our selected region. This is always 1Mb in human, but the fixed size of this view varies between species. This view allows you to scroll back and forth along the chromosome.
You can also drag out and jump to or mark a region.
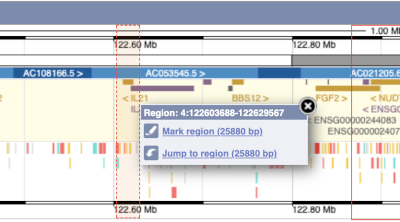
Click on the X to close the pop-up menu.
Click on the Drag/Select button  to change the action of your mouse click. Now you can scroll along the chromosome by clicking and dragging within the image. As you do this you’ll see the image below grey out and two blue buttons appear. Clicking on Update this image would jump the lower image to the region central to the scrollable image. We want to go back to where we started, so we’ll click on Reset scrollable image.
to change the action of your mouse click. Now you can scroll along the chromosome by clicking and dragging within the image. As you do this you’ll see the image below grey out and two blue buttons appear. Clicking on Update this image would jump the lower image to the region central to the scrollable image. We want to go back to where we started, so we’ll click on Reset scrollable image.

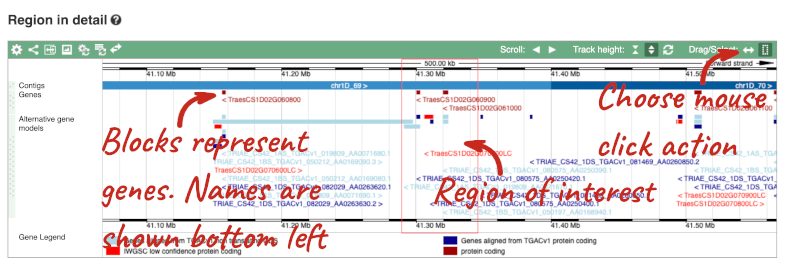
- The third image is a detailed, configurable view of the region.

Here you can see various tracks, which is what we call a data type that you can plot against the genome. Some tracks, such as the transcripts, can be on the forward or reverse strand. Forward stranded features are shown above the blue contig track that runs across the middle of the image, with reverse stranded features below the contig. Other tracks, such as variants, regulatory features or conserved regions, refer to both strands of the genome, and these are shown by default at the very top or very bottom of the view.
You can use click and drag to either navigate around the region or highlight regions of interest, Click on the Drag/Select option at the top or bottom right to switch mouse action. On Drag, you can click and drag left or right to move along the genome, the page will reload when you drop the mouse button. On Select you can drag out a box to highlight or zoom in on a region of interest.
With the tool set to Select, drag out a box around an exon and choose Mark region.
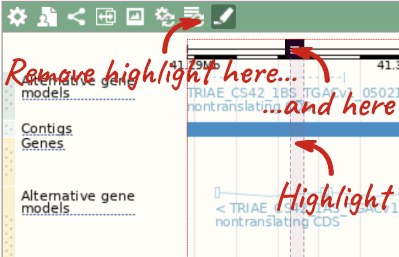
The highlight will remain in place if you zoom in and out or move around the region. This allows you to keep track of regions or features of interest.
We can edit what we see on this page by clicking on the blue Configure this page menu at the left.

This will open a menu that allows you to change the image.
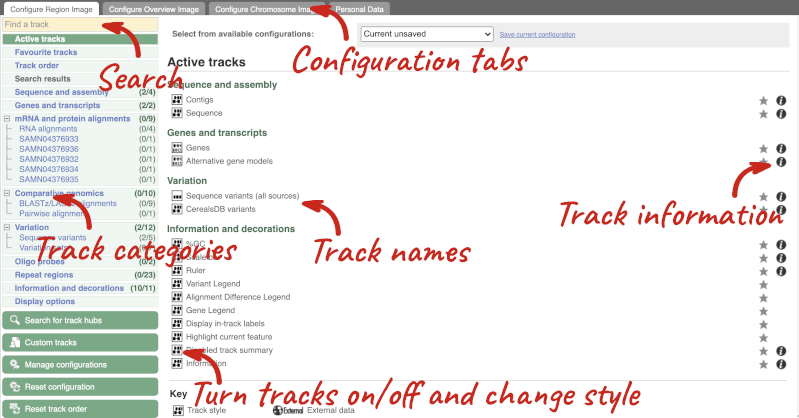
There are thousands of possible tracks that you can add. When you launch the view, you will see all the tracks that are currently turned on with their names on the left and an info icon on the right, which you can click on to expand the description of the track. Turn them on or off, or change the track style by clicking on the box next to the name. More details about the different track styles are in this FAQ.
You can find more tracks to add by either exploring the categories on the left, or using the Find a track option at the top left. Type in a word or phrase to find tracks with it in the track name or description.
Let’s add some tracks to this image. Add:
- EMS-induced mutation variants
- Type I Transposons/LINE (Repeats: Repbase)
Now click on the tick in the top left hand to save and close the menu. Alternatively, click anywhere outside of the menu. We can now see the tracks in the image.
If the track is not giving you can information you need, you can easily change the way the tracks appear by hovering over the track name then the cog wheel to open a menu. To make it easier to compare information between tracks, such as spotting overlaps, you can move tracks around by clicking and dragging on the bar to the left of the track name.

Now that you’ve got the view how you want it, you might like to show something you’ve found to a colleague or collaborator. Click on the Share this page button to generate a link. Email the link to someone else, so that they can see the same view as you, including all the tracks you’ve added. These links contain the Ensembl release number, so if a new release or even assembly comes out, your link will just take you to the archive site for the release it was made on.

To return this to the default view, go to Configure this page and select Reset configuration at the bottom of the menu.
Due to hybridisations in wheat’s evolutionary history, it has a hexaploid genome with related homoeologous regions. We can compare these with the Polyploid view. First, let’s zoom in on the gene TraesCS1D02G061000 by dragging out a box around it and clicking on Jump to region. Now click on the Polyploid view link in the left-hand menu.
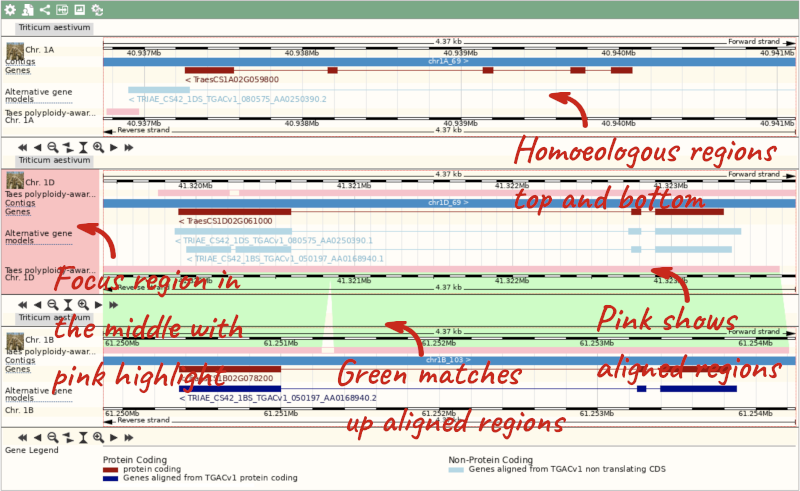
This view also allows us to configure the page, as we could with the main region view, so that we can compare other features between the homoeologous chromosomes.
Exploring a genomic region in Oryza sativa Japonica (rice)
Go to the Ensembl Plants homepage and do the following:
-
Go to the region between 405000 and 453000 on chromosome 1 in Oryza sativa Japonica.
-
Turn on the AGILENT:G2519F-015241 microarray track. Are there any oligo probes that map to this region?
-
Highlight the region around any reverse strand probes you can see. Do they map to any Ensembl transcripts?
-
Go to the Ensembl Plants homepage. Select Oryza sativa Japonica from the Species drop-down list and type
1:405000-453000. Click Go. - Click on Configure this page to open the menu. You can find the AGILENT:G2519F-015241 track under Oligo probes in the left-hand menu, or by using the Find a track box at the top right. Turn on the track as Normal then save and close the menu. As the AGILENT:G2519F-015241 track is stranded, it appears at the top and bottom of the view.
There are 5 probes mapped to this region on the positive strand and one probe on the reverse strand.
- Drag a box around the reverse strand probe then click on Mark region to highlight.
The highlighted region maps to two transcripts: Os01t0107900-02 and Os01t0107900-01
Exploring a wheat region
-
Go to 2D:378720500-378780600 in Triticum aestivum (wheat).
-
How many genes are in this region? What strand are the genes on? What are the gene IDs for these genes?
-
What tracks can you see that show gene structure? Where did the different tracks come from?
-
Export the genomic sequence for this region.
-
Can you view the genomic alignments of the homoeologous regions? What are the different formats you can export the image as?
-
Go to the Ensembl Plants homepage. Select Search: Triticum aestivum and type
2D:378720500-378780600in the text box. Click Go. -
There are two genes displayed in the Genes track. They are both located on the reverse strand. The IDs are
-
There are two tracks which have mapping to this gene: Genes and Alternative gene models. Click the track names for more information on their source.
-
Click Export data in the left-hand menu. Leave the default parameters as they are. Click Next>. Click on Text. Note that the sequence has a header that provides information about the genome assembly, the chromosome, the start and end coordinates and the strand. For example:
>2D dna:chromosome chromosome:IWGSC:2D:378720500:378780600:1 -
Click on Polyploid view in the left hand menu to view the homoeologous regions. Click on Export image. This will open a pop-up menu of the different image formats you can export, which are PNG and PDF.
Exploring a genomic region in coffee
Go to Ensembl Plants.
-
Go to the region from 23,704,000 to 23,766,000 bp on coffee chromosome 1.
-
Zoom in on the GSCOC_T00030044001 gene with transcript ID CDP09644.
-
Configure this page to turn on the Repeats (Repbase) track in this view. What tool was used to annotate the repeats according to the track information? How many repeats can you see within the GSCOC_T00030044001 gene? Do any overlap exons?
-
Create a Share link for this display. Email it to your neighbour. Open the link they sent you and compare. If there are differences, can you work out why?
-
Export the genomic sequence of the region you are looking at in FASTA format.
-
Turn off all tracks you added to the Region in detail page.
-
Go to the Ensembl Plants homepage, select Coffea canephora from the Species drop-down list and type
1:23704000-23766000in the text box. Click Go. -
Draw with your mouse a box encompassing the GSCOC_T00030044001 transcript (with ID CDP09644). Click on Jump to region in the pop-up menu.
- Click Configure this page in the side menu (or on the cog wheel icon in the top left hand side of the bottom image). Go into Repeat regions in the left-hand menu then select Repeats (Repbase). Click on the (i) button to find out more information.
Repeats identified by RepeatMasker, using the Repbase library of repeat profiles.
Save and close the new configuration by clicking on ✓ (or anywhere outside the pop-up window). There are no repeats from Repbase overlapping GSCOC_T00030044001. -
Click Share this page in the side menu. Copy the URL. Get your neighbour’s email address and compose an email to them, paste the link in and send the message. When you receive the link from them, open the email and click on your link. You should be able to view the page with the new configuration and data tracks they have added to in the Location tab. You might see differences where they specified a slightly different region to you, or where they have added different tracks.
-
Click Export data in the side menu. Leave the default parameters as they are (FASTA sequence should already be selected). Click Next>. Click on Text. Note that the sequence has a header that provides information about the genome assembly, the chromosome, the start and end coordinates and the strand. For example:
>1 dna:chromosome chromosome:AUK_PRJEB4211_v1:1:23755890:23764847:1 - Click Configure this page in the side menu. Click Reset configuration. Click ✓.
Exploring a genomic region in Gallus gallus (Chicken)
-
Go to the region from 38,111,022-38,265,293 on chicken chromosome 5. How many contigs make up this portion of the assembly (contigs are contiguous stretches of DNA sequence that have been assembled solely based on direct sequencing information)?
-
Zoom in on ESRRB gene.
-
Turn on the RefSeq GFF3 annotation track as Expanded with labels.
-
Save this image in PDF format.
- Go to the Ensembl homepage. Select Chicken from the drop-down menu in the blue box and enter 5:38111022-38265293 into the text box. Click Go.
This genomic region is made up of one contig indicated by the dark blue coloured bar in the Contigs track.
-
Make sure your cursor is set to the Select a region action (you can change your cursor action in the top right-hand corner of the Region in detail view). Drag a box around the ESRRB gene (note that you will need to highlight the feature itself, i.e. the block, rather than the label) and click on Jump to region.
-
Click on Configure this page in the left-hand panel to open the configuration menu. Enter RefSeq GFF3 annotation into the search box in the top left-hand corner. To enable the track, click on the square next to the track name RefSeq GFF3 annotation and select the Expanded with labels style. Save and close the pop-up menu.
- Click on the Export this image icon above the image and then on the Download button to download the image in PDF format.
Exploring a genomic region in Sus scrofa (Pig)
-
Go to the region from 8,805,953 to 8,858,418 on pig chromosome 11.
-
Configure this page to turn on the Tandem repeats (TRF) track in this view. What is this track? How many TRF overlap this region?
-
Create a URL for this display. Email it to your neighbour.
-
Export the genomic sequence of the region you are looking at in FASTA format.
-
Turn off all tracks you added to the Region in detail page.
-
Go to the Ensembl homepage. Select Pig from the drop-down menu in the blue box and enter
11:8,805,953-8,858,418in the text box. Click Go. - Click Configure this page in the left-hand menu (or on Add/remove tracks at the top left-hand corner of the Region in detail image). Type TRF into the search field in the top left-hand corner of the pop-up menu. Enable the Tandem repeats (TRF) track on the right. You can click on the i icon on the far left for a track description.
The TRF track locates adjacent copies of a pattern of nucleotides. Save and close the new configuration by clicking on the check icon in the top right-hand corner of the pop-up menu or by clicking anywhere outside the pop-up menu. There are 19 TRF that overlap this region.
-
Click Share this page in the left hand-side panel. Copy the URL, get your neighbour’s email address and send them the URL you copied. When you receive the link from them, open the email and click on your link. You should be able to view the page with the new configuration and data tracks they have added.
-
Click on Export data in the left-hand menu. Leave the default parameters as they are. Click Next> and view the sequence in a new browser tab by clicking on Text. The sequence is in FASTA formatwhich comprises a header (beginning with >) that provides information about the genome assembly (primary_assembly:Sscrofa11.1), the chromosome, the start and end coordinates and the strand. For example:
>primary_assembly:Sscrofa11.1:11:8805953-8858418:1 - Click on Reset configuration at the top of the Region in detail image.
Exploring a genomic region in Ovis aries Texel (Sheep)
-
Go to the region 18:7146000-7409000 in the Texel Sheep genome. What genes are found in this region? What strand are they on?
-
Zoom into the start of the first exon of the gene on the left. Zoom in until you can see the genome sequence as coloured bases.
-
Turn on the tracks for translated sequence and start/stop codons. Can you find the start codon? What does this tell you about the gene?
- Go to the Ensembl homepage. Click on View full list of all species. Use the filter in the top right-hand corner of the table to search for Sheep. Click on Sheep (texel) from the list of genomes to open the species information page. From there, search for 18:7146000-7409000 and click Go.
There are three genes in this region, ENSOARG00000010005 and ENSOARG00000010107 on the forward strand, and ENSOARG00000010101 on the reverse strand.
-
Make sure that your cursor action is set to Select a region. with your cursor, drag a box around the start of the first exon of the ENSOARG00000010005 gene, at the left of the view. Click on Jump to region in the pop-up window to zoom in. If you have not zoomed in far enough, drag out another box around the first exon and click on Jump to region. The nucleotide sequence will appear either side of the blue contig as pale blue (C), yellow (G), green (A) and pink (T) boxes. As you zoom in further, you will see the letters on the bases.
- Click on Configure this page and click on Sequence and assembly. Turn on the tracks for Translated sequence and Start/stop codons. Alternatively, you can find the tracks by typing their names into the search field in the top left-hand corner. Close the menu. You can now see the amino acid sequence in all three frames on both strands above and below the nucleotide sequence. Start and stop codons are highlighted either side of these. Start codons are shown in green and stop codons in red.
There is no start codon or methionine residue at the 5’ end of this gene. This suggests that this gene model is incomplete.
Exploring a genomic region in Rainbow trout
-
Go to the region from 49,000,000 to 49,400,000 bp on Rainbow trout chromosome 3.
-
Zoom in on the myo3b gene.
-
Configure this page to turn on the CpG islands track in this view. What tool was used to annotate the CpG islands according to the track information? How many CpG islands can you see within the myo3b gene?
-
Create a Share link for this display. Email it to your neighbour. Open the link they sent you and compare. If there are differences, can you work out why?
-
Export the genomic sequence of the region you are looking at in FASTA format.
-
Turn off all tracks you added to the Region in detail page.
-
Go to the Ensembl homepage. Select Rainbow trout from the Species drop-down list and type
3:49000000-49400000in the text box. Click Go. -
Draw with your mouse a box encompassing the myo3b transcripts. Click on Jump to region in the pop-up menu.
- Click Configure this page in the side menu (or on the cog wheel icon in the top left hand side of the bottom image). Go into Simple features in the left-hand menu then select CpG islands. Click on the (i) button to find out more information.
The CpG islands are determined from the genomic sequence using a program written by G. Micklem, similar to newcpgreport in the EMBOSS package. Save and close the new configuration by clicking on ✓ (or anywhere outside the pop-up window). There is one CpG island overlapping myo3b.
-
Click Share this page in the side menu. Copy the URL. Get your neighbour’s email address and compose an email to them, paste the link in and send the message. When you receive the link from them, open the email and click on your link. You should be able to view the page with the new configuration and data tracks they have added to in the Location tab. You might see differences where they specified a slightly different region to you, or where they have added different tracks.
-
Click Export data in the side menu. Leave the default parameters as they are (FASTA sequence should already be selected). Click Next>. Click on Text. Note that the sequence has a header that provides information about the genome assembly (USDA_OmykA_1.1), the chromosome, the start and end coordinates and the strand. For example:
>3 dna:primary_assembly primary_assembly:USDA_OmykA_1.1:3:49195318:49199101:1 - Click Configure this page in the side menu. Click Reset configuration. Click ✓.
Genes and Transcripts
Demo: Viewing genes and transcripts
You can find out lots of information about Ensembl genes and transcripts using the browser. If you’re already looking at a region view, you can click on any transcript and a pop-up menu will appear, allowing you to jump directly to that gene or transcript.

Alternatively, you can find a gene by searching for it. You can search for gene names or identifiers, and also phenotypes or functions that might be associated with the genes.
We’re going to look at the Triticum aestivum TraesCS3D02G007600 gene. From the Ensembl Plants homepage, type TraesCS3D02G007600 into the search bar and click the Go button.
The gene tab
Click on TraesCS3D02G007600 from the search hits. The Gene tab should open:
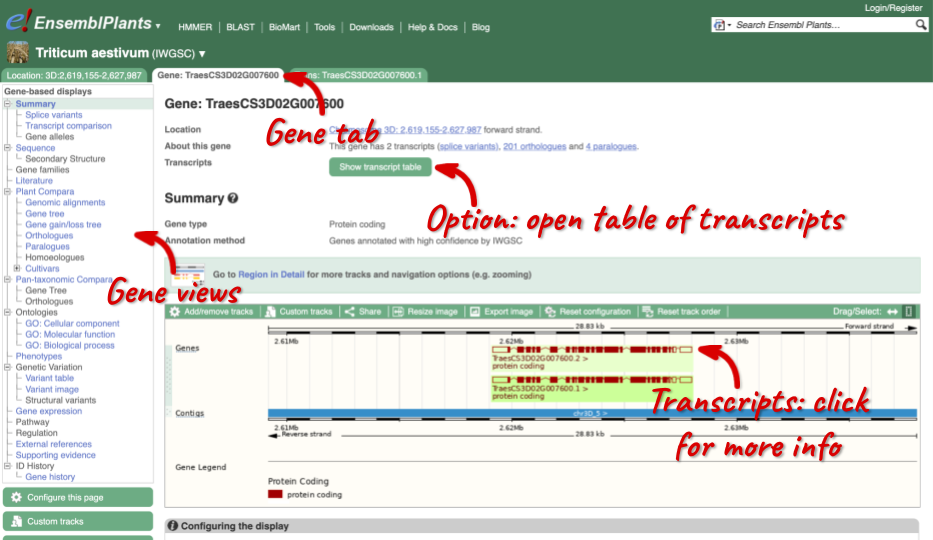
This page summarises the gene, including its location, name and equivalents in other databases. At the bottom of the page, a graphic shows a region view with the transcripts. We can see exons shown as blocks with introns as lines linking them together. Coding exons are filled, whereas non-coding exons are empty. We can also see the overlapping and neighbouring genes and other genomic features.
There are different tabs for different types of features, such as genes, transcripts or variants. These appear side-by-side across the blue bar, allowing you to jump back and forth between features of interest. Each tab has its own navigation column down the left hand side of the page, listing all the things you can see for this feature.
Gene sequence
Let’s walk through this menu for the gene tab. How can we view the genomic sequence? Click Sequence at the left of the page.
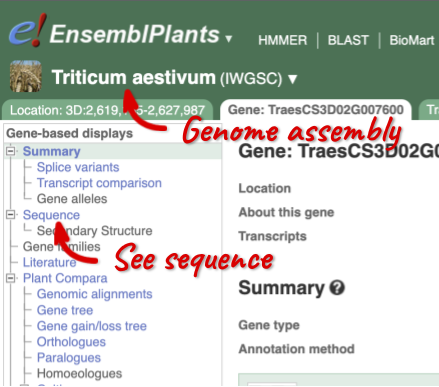
The sequence is shown in FASTA format. The FASTA header contains the genome assembly, chromosome, coordinates and strand (1 or -1) – this gene is on the positive strand.
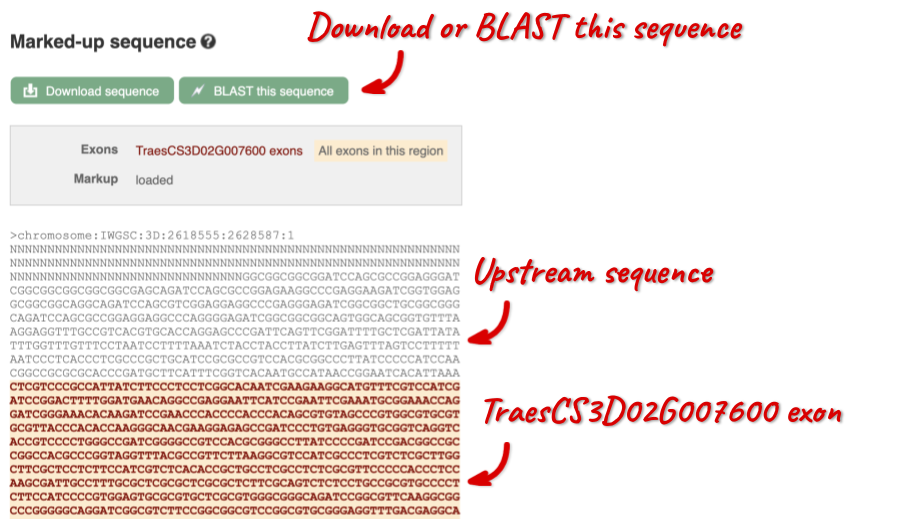
Exons are highlighted within the genomic sequence, both exons of our gene of interest and any neighbouring or overlapping gene. By default, 600 bases are shown up and downstream of the gene. We can make changes to how this sequence appears with the blue Configure this page button found at the left. This allows us to change the flanking regions, add variants, add line numbering and more. Click on it now.
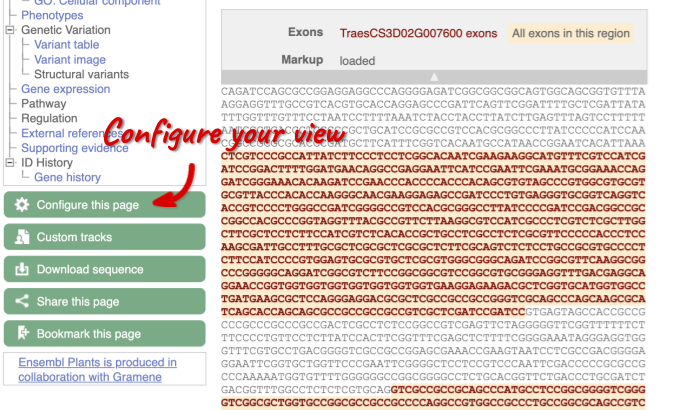
Once you have selected changes (in this example, Show variants and Line numbering) click at the top right.

You can download this sequence by clicking in the Download sequence button above the sequence:
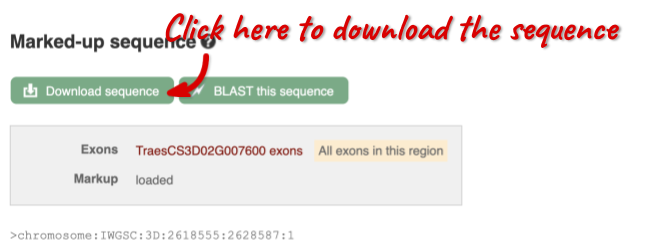
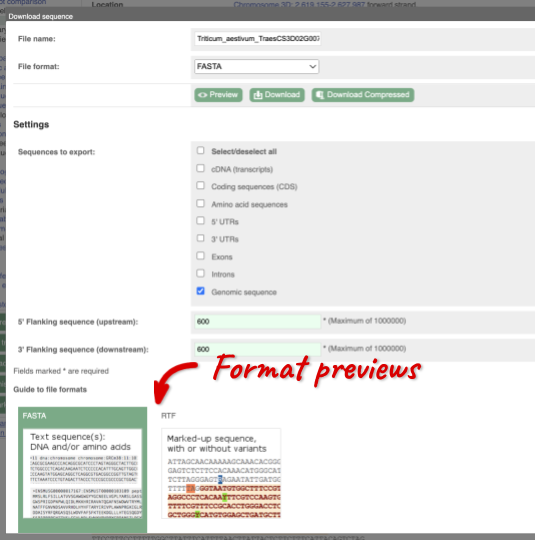
This will open a dialogue box that allows you to pick between plain FASTA sequence, or sequence in rich-text format (RTF), which includes all the coloured annotations and can be opened in a word processor. If you want run a sequence analysis tool, download as FASTA sequence, whereas if you want to analyse the sequence visually, RTF is best for this. This button is available for all sequence views.
Gene function
To find out what the protein does, have a look at GO terms from the Gene Ontology consortium. There are three pages of GO terms, representing the three divisions in GO: Biological process (what the protein does), Cellular component (where the protein is) and Molecular function (how it does it). Click on GO: Biological process to see an example of the GO pages.

Here you can see the functions that have been associated with the gene. There are three-letter codes that indicate how the association was made, as well as links to the specific transcript they are linked to.
Gene information in external databases
We also have links out to other databases which have information about our genes and may focus on other topics that we don’t cover, like Expression Atlas or UniProtKB. Go up the left-hand menu to External references:
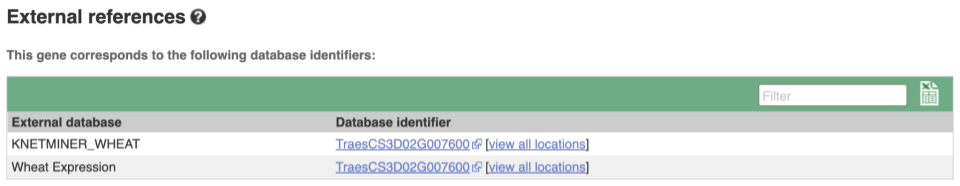
The transcript tab
We’re now going to explore the different transcripts of TraesCS3D02G007600. Click on Show transcript table at the top.
![]()
Here we can see a list of all the transcripts of TraesCS3D02G007600 with their identifiers, lengths and biotypes. Click on the ID of the Ensembl Canonical transcript, TraesCS3D02G007600.2.
You are now in the Transcript tab for TraesCS3D02G007600.2. We can still see the gene tab so we can easily jump back. The left hand navigation column provides several options for the transcript TraesCS3D02G007600.2 - many of these are similar to the options you see in the gene tab, but not all of them. If you can’t find the thing you’re looking for, often the solution is to switch tabs.
Transcript sequences
Click on the Exons link. This page is useful for designing RT-PCR primers because you can see the sequences of the different exons and their lengths.
![]()
You may want to change the display (for example, to show more flanking sequence, or to show full introns). In order to do so click on Configure this page and change the display options accordingly.
Now click on the cDNA link to see the spliced transcript sequence with the amino acid sequence. This page is useful for mapping between the RNA and protein sequences, particularly genetic variants.
![]()
UnTranslated Regions (UTRs) are highlighted in dark yellow, codons are highlighted in light yellow, and exon sequence is shown in black or blue letters to show exon divides. Sequence variants are represented by highlighted nucleotides and clickable IUPAC codes are above the sequence.
Transcript information in external databases
Next, follow the General identifiers link at the left. Just like the External References page in the gene tab, this page shows links out to other databases such as RefSeq, UniProtKB, PDBe and others, this time linked to the transcript or protein product, rather than the gene.
![]()
Protein domain information
If you’re interested in protein domains, you could click on Protein summary to view domains from Pfam, PROSITE, Superfamily, InterPro, and more. These are all plotted against the transcript sequence, with the exons shown in alternating shades of purple at the top of the page. Alternatively, you can go to Domains & features to see a table of the same information.
![]()
![]()
Exploring a Triticum aestivum (Wheat) gene
Start in the Ensembl Plants homepage and select the Triticum aestivum IWGSC genome to answer the following questions:
-
What GO: Molecular function terms are associated with the Wheat gene TraesCS6D02G180200?
-
Go to the transcript tab. How many exons does it have? Which one is the longest? Approximately, how much of that is coding?
-
What domains can be found in the protein product of this transcript? What prediction method(s) identified these domains?
- Go to Ensembl Plants, select Triticum aestivum from the drop down menu then type
TraesCS6D02G180200into the search box. Click on the gene name link TraesCS6D02G180200 in the search results. Click on GO: Molecular function in the left-hand menu.There is one term listed: GO:0005515, protein binding.
- Click on the transcript tab at the top of the page. Click on Exons in the left-hand menu.
There are six exons. Exon 6 is longest with 485 bp, of which around one sixth is coding.
- Click on either Protein Summary or Domains & features in the left-hand menu to view the data graphically or as a table, respectively.
Leucine-rich repeats are predicted by many different methods, however each method predict the leucine-rich repeats at different positions.
Exploring a defence-related gene in Tomato, Solanum lycopersicum
(a) Search for the tomato gene NCED2 and go to the gene tab.
- What is the amino acid length of the only transcript of this gene?
- Which chromosome and which strand of the genome is this gene located?
(b) Look at the gene Description field, what does this tell you about the cellular localisation of the protein product of this gene? Does this match the Gene Ontology (GO): Cellular component terms? Click on GO:Cellular component to check.
(c) Click on Gene expression. Which tissue has the highest expression of this gene according to the Tomato Genome Consortium?
(d) The summary at the top of the page (just above the Show transcript table button) shows us that there are nine paralogues of this gene. Click on the Gene gain/loss tree to look at the expansion of this gene family across all plants.
- Which species has the largest number of members of this gene family?
(e) Go to the transcript tab for this gene by clicking on the transcript ID Solyc08g016720.1.1 from the transcript table. Are there any Oligo probes that would be useful in targeting this gene experimentally?
(a) Go to plants.ensembl.org and type NCED2 into the search box, selecting Solanum locypersicum from the drop down menu. Click on the first result to go to the gene tab.
Click on the Show transcript table button if the transcript table is hidden. In the 4th column we see the protein length listed, 581 amino acids in length.
The location is listed at the top of the page, we can see that this is on Chromosome 8, between the base pairs 8,729,953 and 8,731,698, and on the forward strand.
(b) The gene description for this gene is ‘9-cis-epoxycarotenoid dioxygenase NCED2, chloroplastic’ which suggests the enzyme is localised to the chloroplast.
In the left-hand navigation panel, find the link to GO: Cellular location. We can see three results, chloroplast, plastid and chloroplast stroma, so this matches the gene description.
(c) Click on Gene expression in the left-hand navigation panel.
Darker shades of blue indicate higher expression. Hover your mouse over the heat-map to show a pop-up with the TPM (Transcripts Per Kilobase Million).
The 2cm fruit in the Tomato Genome Consortium has the highest expression at 103 TPM. You can also click on Filters at the top right and filter to high or medium expression.
(d) Click on the Gene gain/loss tree. You might find it easier to compare in the radial tree, click the two arrows icon at the top left of the image () to toggle to the radial view.
Look for the red lines, indicating the larger number of members and significant expansion. The number of members are listed just before the species name.
Brassica napus and Brassica juncea has the highest number of members in this gene family.
(e) Go to the transcript tab for this gene by clicking on the transcript ID Solyc08g016720.1.1 from the transcript table.
Find the Oligo probes link in the left-hand navigation panel. There is a single probe from Affymetrix, the AFFY TomGene, 20363698.
Exploring the FUS3 gene in Coffea canephora (Robusta coffee)
The FUS3 gene is a known master regulator of somatic embryogenesis, an important factor in stable genetic transformation and successful plant regeneration of coffee trees expressing the Bacillus thuringiensis (Bt) toxin Cry10Aa to induce Coffee Berry Borer (CBB) resistance.
-
Find the Coffea canephora FUS3 gene on Ensembl Plants. On which chromosome and which strand of the genome is this gene located?
-
Where in the cell is the FUS3 protein located?
-
What is the source of the assigned gene description?
-
How long is its transcript (in bp)? How long is the protein it encodes? How many exons does it have? Are any of the exons completely or partially untranslated?
- Go to the Ensembl Plants homepage (http://plants.ensembl.org/). Select C. canephora from the species list and type
FUS3in the search box. Click Go and click on the gene ID GSCOC_T00019208001. You can find the strand orientation and the location under Summary in the Gene tab.The C. canephora FUS3 gene is located on chromosome 7 on the forward strand.
- Click on GO: Cellular component in the left-hand panel.
The protein is located in the nucleus.
- Click on Summary in the side menu.
The gene description is Projected from Arabidopsis thaliana (AT3G26790) by UniProtKB/Swiss-Prot;Acc:Q9LW31.
- Click on Show transcript table.
The transcript is 1038 bp and the length of the encoded protein is 279 amino acids.
Click on the transcript ID CDP16731 in the transcript table. You can find the number of exons in under in the summary information at the top of the page.
It has 7 exons.
Click on Sequence: Exons in the left-hand panel.
The last exon is partially untranslated (sequence shown in orange). This can also been seen from the fact that in the transcript diagrams on the Gene Summary and Transcript Summary pages the boxes representing the last exon is partially unfilled.
Exploring a bacterial gene in Clostridium sporogenes
Start in Ensembl Bacteria and select the Clostridium sporogenes (GCA_001444695) genome.
-
What is the gene name for the Glutamine synthetase gene?
-
Go to the transcript tab. How long is the transcript? How long is the protein?
-
What domains can be found in the protein product of this transcript? How many different domain prediction methods agree with each of these domains?
- From the Ensembl Bacteria homepage, select Clostridium sporogenes by beginning to write the species name and selecting the species from the auto-complete list. Type
Glutamine synthetaseand click on the gene ID ENSB:yZtlLO8Ti90y75J which will open the Summary display on the Gene tab..The gene name is
glnA. - Switch to the Transcript tab and go to the Summary display. You can find the length under Statistics underneath the transcript image.
The
glnAtranscript is 1,899 bp and the protein is 632 aa in length. - Click on either Protein Summary or Domains & features in the left hand menu to see graphically or as a table respectively.
6 protein domains were found. All of them predict a glutamine synthetase domain.
Exploring the MYH9 gene in Gallus gallus (Chicken)
- Find the MYH9 (myosin, heavy chain 9, non-muscle) gene in the chicken reference, and go to the Gene tab.
- On which chromosome and which strand of the genome is this gene located?
- Which transcript produces the longest protein and how long is the protein sequence?
-
What are some functions of MYH9 according to the Gene Ontology consortium? Have a look at the GO pages for this gene.
- In the transcript table, click on the transcript ID for MYH9-209, and go to the Transcript tab.
- How many exons does it have?
- Are any of the exons completely or partially untranslated?
- Is there an associated sequence in UniProt? Have a look at the General identifiers for this transcript.
- Are there microarray (oligo) probes that can be used to monitor ENSGALT00010036169.1 expression?
- Go to the Ensembl homepage. Select Chicken from the drop-down list in the blue box, enter MYH9 and click Go. In the search results page, click on Chicken reference in the left-hand panel to restrict your results to the reference genome only. Click on the first hit MYH9 (Chicken Gene, Breed: reference) to open the Gene tab. Look at the Location section in the gene summary at the top of the page.
The gene is located on chromosome 1 on the forward strand.
Now click on the Show transcript table button and focus on the Protein column in the Transcript table.
The transcript ENSGALT00010036169.1 (MYH9-209) produces the longest protein at 1,960 amino acid residues.
- Gene Ontology maps terms to a protein in three classes: biological process, cellular component, and molecular function.
Meiotic spindle organisation, cell morphogenesis, and angiogenesis are some of the roles associated with the MYH9 gene.
- Click on ENSGALT00010036169.1 in the Transcript table to open the corresponding Transcript tab. Look at the About this transcript section in the transcript summary at the top of the page.
The transcript has 41 exons.
Click on the Exons link in the left-hand side menu. In the Sequence column of the Exon table, look for any UnTranslated Regions (UTRs) which coloured in orange.
Exon 1 is completely untranslated, and exons 2 and 41 are partially untranslated. You can also see this in the cDNA view if you click on Sequence: cDNA in the left-hand menu.
Click on External References: General identifiers in the left-hand menu. Look for UniProtKB in the External database column.
A0A1D5PM19.34 from UniProt matches the translation of the Ensembl transcript. Click on A0A1D5PM19.34 to open the corresponding UniProt entry in a new browser tab.
- In the left-hand menu, look for External References: Oligo probes.
There are probes from Affy and Agilent that can be used to monitor expression of this transcript.
Exploring the Sus scrofa (Pig) TRAF3 gene
- Find the TRAF3 (TNF receptor associated factor 3) gene in the pig reference, and go to the Gene tab.
- Which strand of the genome is this gene located?
- How many transcripts are there of the gene?
- Which transcript produces the longest protein and how long is the protein sequence?
-
What are some functions of TRAF3 according to the gene ontology (GO)? Have a look at the Ontologies pages for this gene.
- In the transcript table, click on the transcript ID for TRAF3-201, and to open the corresponding Transcript tab.
- How many exons does it have?
- Are any of the exons completely or partially untranslated?
- Is there an associated sequence in UniProt? Have a look at the General identifiers for this transcript.
-
Are there microarray (oligo) probes that can be used to monitor the expression of TRAF3-201?
- Now find the TRAF3 gene in the Berkshire pig breed.
- Which strand of the genome is this gene located?
- How many transcripts are there of the gene?
- Which transcript produces the longest protein and how long is the protein sequence?
- How do the Ensmebl canonical transcripts differ between the pig reference and the Berkshire breed?
- Go to the Ensembl homepage. Select Pig from the drop-down list in the blue box, enter TRAF3 into the text box and click Go. In the search results page, click on Pig reference in the left-hand panel to restrict your results to the pig reference only. Click on the first hit TRAF3 (Pig Gene, Breed: reference) to open the Gene tab. Look at the Location section in the gene summary at the top of the page.
The TRAF3 gene is located on the forward strand.
Now look at the About this gene section in the gene summary at the top of the page.
TRAF3 has 4 transcripts.
Click on the Show transcript table button underneath the gene summary. Focus on the Protein column in the transcript table.
The transcript ENSSSCT00000101908.1 (TRAF3-201) produces the longest protein at 573 amino acid residues in length.
- Gene Ontology maps terms to a protein in three classes: biological process, cellular component, and molecular function.
Some of the GO terms associated to the TRAF3 gene are: regulation of cytokine production and proteolysis (biological process), protein kinase and metal ion binding (molecular function), and cytoplasm and endosome (cellular component).
- Click on ENSSSCT00000101908.1 in the transcript table. Under the summary information at the top of the page, focus on the About this transcript section.
This transcript has 11 exons.
Click on the Exons link in the left-hand side menu. In the Sequence column of the Exon table, look for any UnTranslated Regions (UTRs) which coloured in orange.
Only exon 11 is partially untranslated. You can also see this in the cDNA view if you click on Sequence: cDNA in the left-hand menu.
Click on External References: General identifiers in the left-hand menu. Look for UniProtKB in the External database column.
A0A4X1TTD0.19 and A0A8W4FAU5.6 from UniProt match the translation of the Ensembl transcript. Click on the IDs to open the corresponding entry in UniProt.
- In the left-hand menu, look for External References: Oligo probes.
The link is greyed out, which means that no commercial oligo probes are available to monitor expression of this transcript.
- Go to the Ensembl homepage by clicking on the Ensembl logo in the top left-hand corner of any page. Select Pig from the drop-down list in the blue box, enter TRAF3 into the text box and click Go. In the search results page, click on Pig berkshire in the left-hand panel to restrict your results to the Berkshire breed only. Click on the first hit TRAF3 (Pig Gene, Breed: berkshire) to open the Gene tab. Look at the Location section in the gene summary at the top of the page.
The TRAF3 gene is located on the forward strand in the Berkshire breed.
Now look at the About this gene section in the gene summary at the top of the page.
TRAF3 has 5 transcripts in the Berkshire breed.
Click on the Show transcript table button underneath the gene summary. Focus on the Protein column in the transcript table.
The transcript ENSSSCT00065038580.1 (TRAF3-205) produces the longest protein at 573 amino acid residues in length.
- The Ensembl canonical transcript in the pig reference genome is 6,677 bases in length. The Ensembl canonical transcript in the Berkshire breed is 6,340 bases in length. This suggests that the TRAF3 Ensembl canonical transcript in the reference has more/larger UTRs compared to the Berkshire breed. To compare, you can open the Sequence: Exons pages in the Transcript tab in both breeds and look at the length of the UTR (coloured in orange).
Variation
Demo: The gene tab
View all variants within a gene sequence
In any of the sequence views shown in the Gene and Transcript tabs, you can view variants on the sequence. You can do this by clicking on Configure this page from any of these views.
Let’s take a look at the Gene sequence view for TraesCS4A02G446800 in wheat. Search for TraesCS4A02G446800 and go to the Sequence view.
If you can’t see variants marked on this view, click on Configure this page and select Show variants: Yes and show links.

Find out more about a variant by clicking on it.
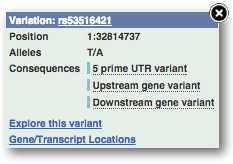
You can add variants to all other sequence views in the same way.
You can go to the Variation tab by clicking on the variant ID. For now, we’ll explore more ways of finding variants.
View all variants within a gene in tabular format
To view all the sequence variations in table form, click the Variant table link at the left of the gene tab
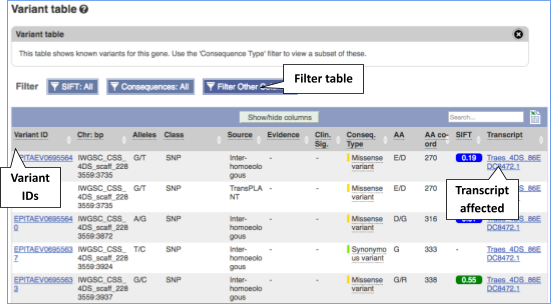
You can filter the table to only show the variants you’re interested in. For example, click on Consequences: All, then select the variant consequences you’re interested in.
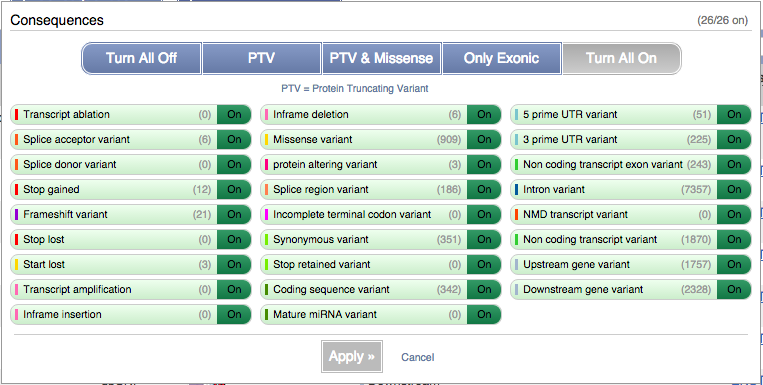
You can also filter by other columns such as, Evidence or Class.
Demo: The location tab
Visualise variants within a region
Let’s have a look at variants in the Location tab. Click on the Location tab in the top bar.
Configure this page and open Variation from the left-hand menu.
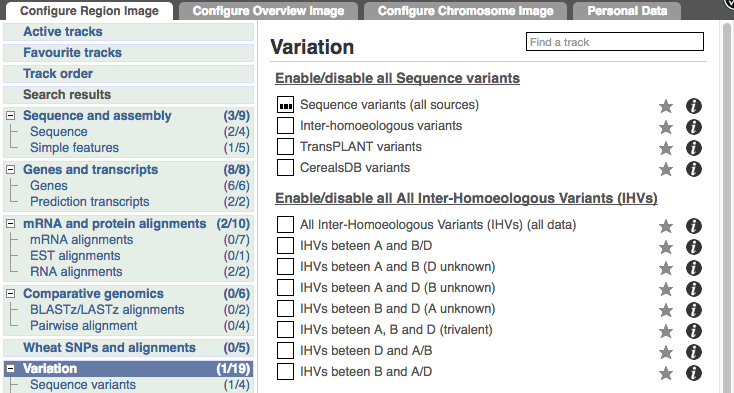
There are various options for turning on variants. You can turn on variants by source, by frequency, presence of a phenotype or by individual genome they were isolated from. Turn on the following all sequence variants in Normal.
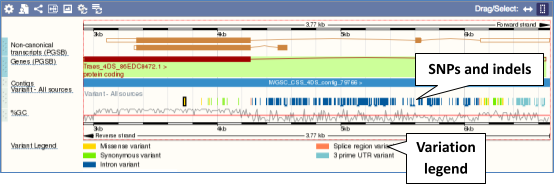
Click on a variant to find out more information. It may be easier to see the individual variants if you zoom in.
Demo: The variant tab
Variant summary
Let’s have a look at a specific variant. The easiest way to find a specific variant is to search for it. Search for BA00249348 and click through to the Variant tab.
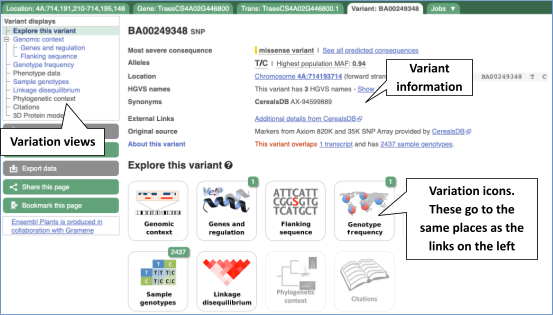
Variant consequences specific features
The icons show you what information is available for this variant. Click on Genes and regulation, or follow the link at the left.
This variant is found in TraesCS4A02G446800 only.
Genotype frequency
Let’s look at population genetics. Either click on Explore this variant in the left hand menu then click on the Genotype frequency icon, or click on Genotype frequency in the left-hand menu.
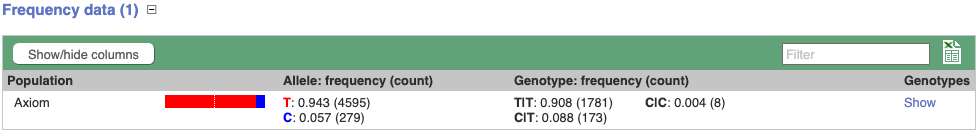
Genotype frequency
We can see which strains these genotypes were observed in by going to Sample Genotypes.

Investigating a variant in wheat
-
Search for the variant
BA00369602in Triticum aestivum on Ensembl Plants. Is this variant known by any other names? -
What gene is affected by this variant? What is the amino acid change?
-
Which cultivars have the alternative base at this locus?
- Start at the homepage and enter BA00369602 into the search box and select Triticum aestivum from the drop-down list. Click on the Gene ID BA00369602 in the search results to get to the variation homepage.
Under Synonyms, you can see that the variant is also known as AX-94448191 in CerealsDB.
- Click on Genes and regulation.
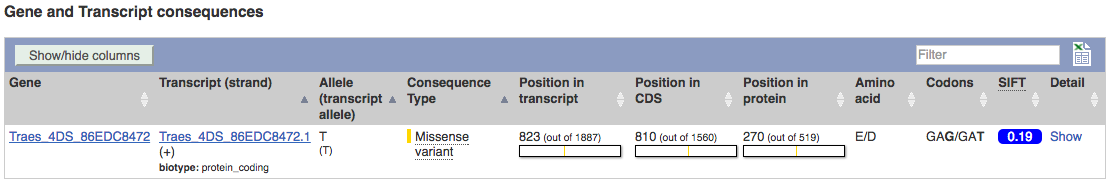
The variant is a missense variant on TraesCS2D02G303800, where it gives a glycine to aspartic acid (G/D) change at transcript position 406.
- Click on Sample genotypes. Scroll down the table to see if there are any cultivars with the A allele in the genotype column.
All of the cultivars listed have the genotype
G|G.
Variation data in Phaseolus vulgaris
-
Go to Ensembl Plants and find the PHAVU_001G219900g gene in Phaseolus vulgaris and go to its Location tab. Can you see the variation track?
-
Zoom in around the first exon of this gene. Are any missense variants mapped in the translated region of this exon?
- Select Phaseolus vulgaris from the Species search drop-down menu and search for
PHAVU_001G219900g. In the results page, you can click on the coordinates 1:48,238,848-48,245,168 to go straight to the Location tab. Scroll down to the Region in detail view. The variation track (phaseolus_vulgaris_eva_PRJEB18671) is shown at the bottom of the view.If you don’t see the Variation - All sources track, click Configure this page on the left-hand panel, search for the track in the pop-up menu and enable the track by clicking on the square next to the track name. Close the pop-up window and wait for the track to load.
- Zoom in around the first exon of this gene by drawing a box in the respective region (you can change your mouse action by clicking the Drag/Select icons at the top right-hand corner of the view). Note the gene is on the reverse strand (this is signified by the < sign next to the transcript name, and it is located below the Contigs track), so the first exon will be on the right hand side of that image. The variation legend is shown at the bottom of the page, telling you what the colours mean.
There are four missense variants within the region; 1:48244305:C_T:PRJEB18671, 1:48244362:T_G:PRJEB18671, 1:48244426:G_A:PRJEB18671, 1:48244435:T_A:PRJEB18671.
Missense variants are shown in yellow. Click on the variants to get additional information on that variant including location. You can zoom into the region if the variant block is too small to click.
The variants are found at 1:48244305, 1:48244362, 1:48244426, 1:48244435.mSNPs are tagged with ambiguity codes (zoom into the region if you cannot see this). You can find a useful IUPAC ambiguity code guid on the bioinformatics.org website.
Variation data in tomato
-
Go to Ensembl Plants and find the Solyc02g084570.3 gene in Solanum lycopersicum (tomato) and go to its Location tab. Can you see the variation track?
-
Zoom in around the last exon of this gene. What are the different types of variants seen in that region? Are any splice region variants mapped in the region? If so, what is/are the coordinate(s)?
- Select Solanum lycopersicum from the Species search drop-down menu and search for
Solyc02g084570.3. In the results page, you can click on the coordinates 2:48284598-48288482 to go straight to the Location tab. Scroll down to the Region in detail view. The variation track is shown at the bottom of the view.If you don’t see the Variation - All sources track, click Configure this page on the left-hand panel, search for the track in the pop-up menu and enable the track by clicking on the square next to the track name. Close the pop-up window and wait for the track to load.
- Zoom in around the last exon of this gene by drawing a box in the respective region (you can change your mouse action by clicking the Drag/Select icons at the top right-hand corner of the view). Note the gene is on the reverse strand (this is signified by the < sign next to the transcript name, and it is located below the Contigs track), so the last exon will be on the left hand side of that image. The variation legend is shown at the bottom of the page, telling you what the colours mean.
The types of variants seen in that region are 3’ UTR, missense, synonymous and splice region variants.
Splice region variants are shown in orange. Click on the variants to get additional information on that variant including location. You can zoom into the region if the variant block is too small to click.
The variants are found at 2:48285642 and 2:48285640-48285641. Note that the two variants overlap: one is a SNP and the other is an indel. SNPs are tagged with ambiguity codes (zoom into the region if you cannot see this). You can find a useful IUPAC ambiguity code guid on the bioinformatics.org website. Single-letter ambiguity codes are given when two or more possible nucleotides may be represented at a single base locus.
Variation data in Fusarium oxysporum
-
How many species in Ensembl Fungi have variation data?
-
Select Fusarium oxysporum (FO2) and search for the FOXG_13574T0 gene. One of its upstream variants is SNP tmp_10_6610. What are the possible alleles for this polymorphic position? Which one is on the reference genome?
-
What is the most frequent allele at this position?
-
Which samples have the genotypes C|T and T|T?
- Go to Ensembl Fungi, click on View full list of all species. You can sort the table by column. Click on the Variation database column to sort the table by species with variation data.
The table shows that we have 8 fungi species currently with variation databases.
- Click on Fusarium oxysporum in the table and on the species page search for
FOXG_13574T0. From the Gene tab, click on Variant table in the left-hand panel. You can use the filter at the top right-hand corner of the tabletmp_10_6610.The alleles are C/T, where C is the reference allele.
- Click on tmp_10_6610 in the table to open the Variant tab. Then click on Genotype frequency from the menu on the left-hand side of the page.
The most frequent allele at this position is C with a frequency of 0.850.
- Click on Sample genotypes in the menu on the left.
The table shows that sample 909454 has the C|T genotype and 909455 has the T|T genotype.
Exploring a SNP in chicken
(a) Find the page with information for the chicken SNP rs10731268.
(b) What gene(s) does rs10731268 fall within? What is its effect?
(c) Have any papers been written mentioning rs10731268? What are they about?
(d) What allele is at this position in other birds? What is the likely ancestral allele?
(a) Go to the Ensembl homepage.
Type rs10731268 in the Search box, then click Go. Click on rs10731268.
(b) Click on Genes and Regulation in the side menu (or the Genes and Regulation icon).
rs10731268 falls within 2 genes: ENSGALG00010028562 and ENSGALG00010028568 (HGNC: MLLT1). This variant has a missense consequence in seven transcripts of the ENSGALG00010028562 gene, and downstream gene variant consequence in three transcripts of the ENSGALG00010028568 (HGNC: MLLT1) gene.
(c) Click on Citations in the left hand side menu.
This variant is mentioned in the paper ‘Identification and characterization of genes that control fat deposition in chickens’ from 2013 by D’Andre et al. Click on the PubMed ID 24206759 to go to the paper.
(d) Click on Phylogenetic Context in the side menu. Select Alignment: 17 sauropsids EPO and click Go.
Japanese quail, Duck, Golden Eagle, Common canary and Zebra finch all have an A in this position. This suggests that A may be the ancestral allele.
Exploring a variant in pig
The human gene MC4R has been associated with obesity. The SNP rs81219178 has been identified as a variant in the pig MC4R gene.
(a) What is the amino acid change caused by rs81219178 in MC4R of the pig? Is the change likely to alter the protein function?
(b) How many transcripts does this variant affect? What are the consequences of this variant?
(a) Go to the Ensembl homepage.
Type rs81219178 in the Search box, then click Go.
Click on rs81219178 (Pig Variant, Breed: reference).
Click on Genes and regulation in the left-hand menu or on the icon.
The variant causes a D->N amino acid change (Aspartic acid -> Asparagine). The SIFT score of 0.01 predicts that this change will have a deleterious effect on the protein.
(b) This variant affects one transcript (ENSSSCT00000091644.1) of ENSSSCG00000051798 gene and it has the missense consequence.
Variation data in sheep CD72
The sheep Cd72 gene product is an integral component of the plasma membrane (GO:0005887).
(a) Can you find all variants that have been described for this gene so far? Do any of them change the amino acid sequence of the protein?
(b) Are any of the missense variants predicted to be deleterious by SIFT? What are their IDs?
(a) Go to the Ensembl homepage.
Select Search: Sheep and type CD72. Click Go.
Click on novel gene (Sheep Gene, Breed: texel) to go to the Gene tab.
In the left-hand menu, click on Variant table to see the full list of variants described for this gene.
Filter the table for: Consequences > missense variant to see which variants cause changes in the amino acid sequence.
There are 23 missense variants.
(b) Click on the heading SIFT of the table to sort the column.
Five missense variants have been predicted to be deleterious by SIFT, shown by the red SIFT scores. Their IDs are: rs593145093, rs1088269383, rs1093464070, rs422891390 and rs597376323.
Exploring a SNP in Atlantic salmon
The missense variant 25:3426821:C_A:PRJEB34225 is found in the Atlantic salmon hs6st2 gene.
(a) Find the page with information for 25:3426821:C_A:PRJEB34225.
(b) Is 25:3426821:C_A:PRJEB34225 a missense variation in all transcripts of the hs6st2 gene?
(c) What is the major allele in 25:3426821:C_A:PRJEB34225?
(a) Please note there is more than one way to get this answer. Either go to the Variant Table for the Atlantic salmon hs6st2 gene, and filter variants to the missense variants, or search Ensembl for 25:3426821:C_A:PRJEB34225 directly.
(b) Once you’re in the Variation tab, click on the Genes and regulation link or icon.
This SNP is found in four transcripts from two genes. It is a missense variant in hs6st2 gene and an intron variant, and downstream gene variant in ENSSSAG00000118991.
(c) Select Population genetics from the side menu.
From the Frequency data table, the PRJEB34225 allele frequencies shows that C is the major allele (95% of all population) compared to A (5% of all population).
VEP
Demonstration of the VEP web interface
Input
We have identified three variants on wheat chromosome 4B:
C -> T at 240206468
C -> G at 240199078
C -> T at 240212229
We will use the Ensembl VEP to determine:
- Have my variants already been annotated in Ensembl?
- What genes are affected by my variants?
- Do any of my variants affect gene regulation?
Click on Tools in the top green bar from any Ensembl Plants page, then Variant Effect Predictor to open the input form:
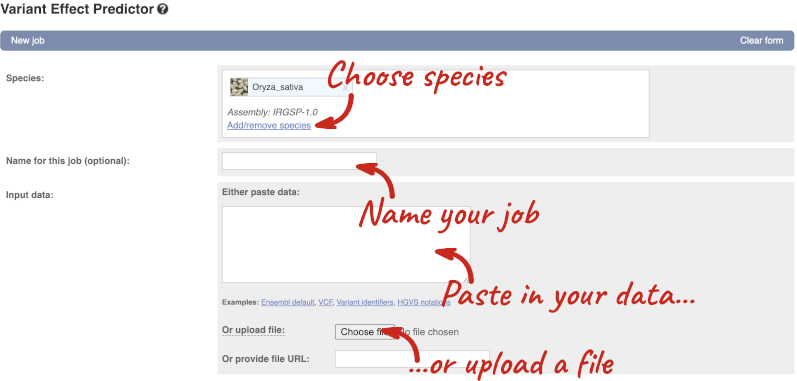
Click on Add/remove species and search for Triticum aestivum to choose it.
The data is in VCF:
chromosome coordinate id reference alternative
Put the following into the Paste data box:
4B 240206468 var1 C T
4B 240199078 var2 C G
4B 240212229 var3 C T
The VEP will automatically detect that the data is in VCF.
Additional configurations
There are further options that you can choose for your output. These are categorised as Identifiers, Variants and frequency data, Additional annotations, Predictions, Filtering options and Advanced options. Let’s open all the menus and take a look.
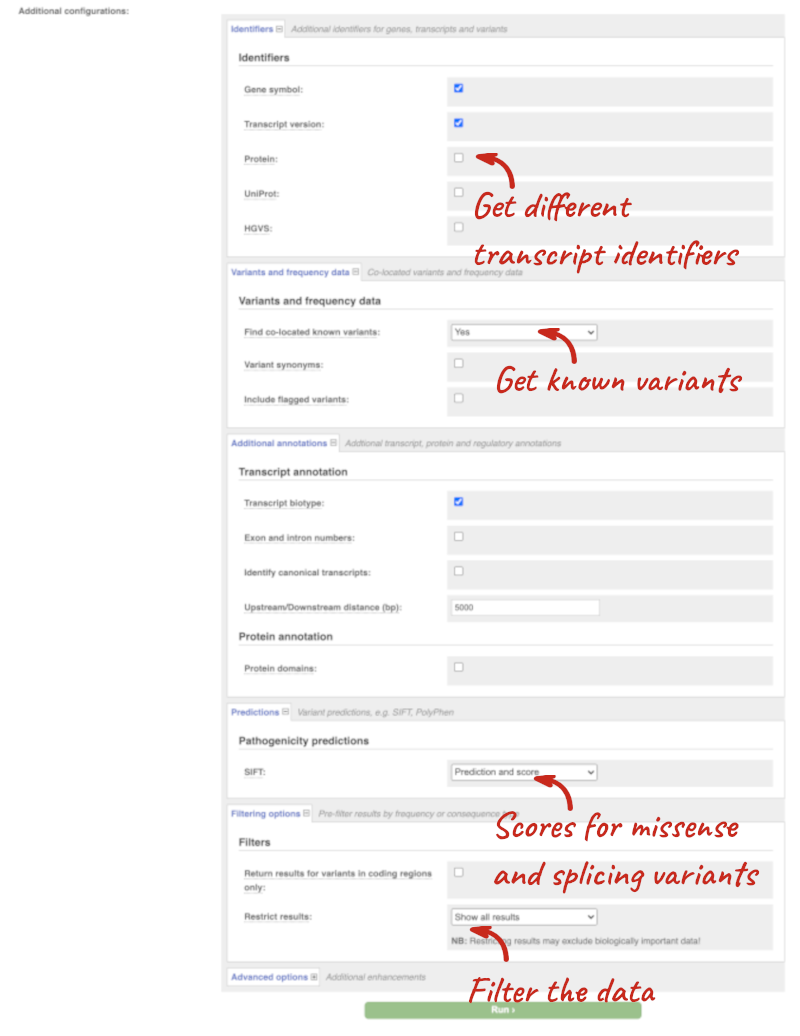
Hover over the options to see definitions.
When you’ve selected everything you need, scroll right to the bottom and click Run.

The display will show you the status of your job. It will say Queued, then automatically switch to Done when the job is done, you do not need to refresh the page. You can edit or discard your job at this time. If you have submitted multiple jobs, they will all appear here.
Click View results once your job is done.
Results
In your results you will see a graphical summary of your data, as well as a table of your results.
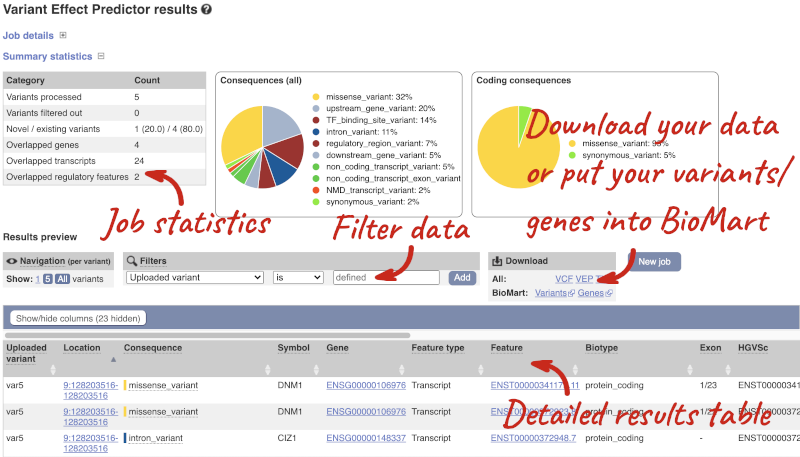
The results table is enormous and detailed, so we’re going to go through the it by section. The first column is Uploaded variant. If your input data contains IDs, like ours does, the ID is listed here. If your input data is only loci, this column will contain the locus and alleles of the variant. You’ll notice that the variants are not neccessarily in the order they were in in your input. You’ll also see that there are multiple lines in the table for each variant, with each line representing one transcript or other feature the variant affects.
You can mouse over any column name to get a definition of what is shown.
The next few columns give the information about the feature the variant affects, including the consequence. Where the feature is a transcript, you will see the gene symbol and stable ID and the transcript stable ID and biotype. The IDs are links to take you to the gene or transcript homepage.
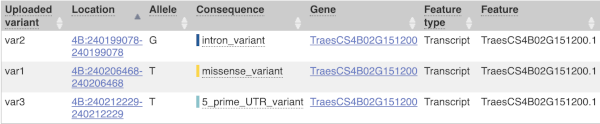
This is followed by details on the effects on transcripts, including the position of the variant in terms of the exon number, cDNA, CDS and protein, the amino acid and codon change and pathogenicity scores. Where the variant is known, the ID of the existing variant is listed, with a link out to the variant homepage. The pathogenicity scores are shown as numbers with coloured highlights to indicate the prediction, and you can mouse-over the scores to get the prediction in words.
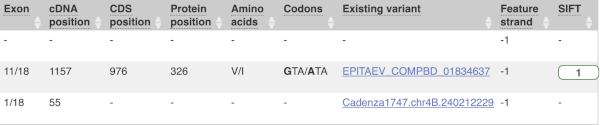
Above the table is the Filter option, which allows you to filter by any column in the table. You can select a column from the drop-down, then a logic option from the next drop-down, then type in your filter to the following box. We’ll try a filter of Consequence, followed by is then missense_variant, which will give us only variants that change the amino acid sequence of the protein. You’ll notice that as you type missense_variant, the VEP will make suggestions for an autocomplete.
You can export your VEP results in various formats, including VCF. When you export as VCF, you’ll get all the VEP annotation listed under CSQ in the INFO column. After filtering your data, you’ll see that you have the option to export only the filtered data. You can also drop all the genes you’ve found into the Gene BioMart, or all the known variants into the Variation BioMart to export further information about them.
Web VEP analysis of variants in Oryza sativa Japonica (rice)
You’ll find a VCF file here. This is a small subset of the outcome of Oryza sativa Japonica whole-genome sequencing and variant-calling experiment. Analyse the variants in this file with the VEP tool in Ensembl Plants and determine the following:
-
How many genes and transcripts are affected by variants in this file?
-
Do these variants result in a change in the proteins encoded by any of the Ensembl genes? Which genes are affected? What is the amino acid change? What is the pathogenicity prediction score for this change?
Go to Ensembl Plants and click on Tools at the top of the page. Click on Variant Effect Predictor and select Oryza sativa Japonica Group from the Species menu.
Either click on Choose file and select the file to upload it, or directly paste the URL into the Or provide file URL: box. Click Run at the bottom of the page. When your job is done, click View results.
- The number of affected genes and transcripts is shown in the Summary statistics table at the top.
8 genes and 8 transcripts are affected by these variants.
- Use the filters to view only missense variants. The filters are found above the detailed results table in the middle. Select Consequence and is from the drop-down menus. Then type
missense_variantinto the boxe. Add to apply your filter.1 variant is a missense variant. It causes a leucine to arginine (L/R) at position 16 change in the gene OS09G0103500. The SIFT score is 0.01 (Deleterious low confidence). Refere to this link for more information on SIFT (https://sift.bii.a-star.edu.sg/).
Web VEP analysis of variants in Triticum aestivum (wheat)
You have done whole-genome sequencing and variant-calling experiments for Triticum aestivum. You have a VCF file with a small subset of variants from this experiment. Analyse the variants in this file with the VEP tool in Ensembl Plants and determine the following:
-
How many variants were analysed? How many are novel?
-
How many genes and transcripts are affected by variants in this file?
-
Do any of the variants have different consequences for different transcripts?
-
Filter the table to find variants with high impact. How many variants have high impact? Why do you think missense variants are not classified as high impact?
-
Can you export all the results to a VCF file? Compare it to the input VCF file to see what information the VEP adds.
Go to any Ensembl Plants page and click on Tools in the navigation bar at the top of the page. Click on Variant Effect Predictor and change your species to Triticum aestivum by clicking on Change species.
Enter a descriptive name for your VEP job. If you have downloaded the variant file to your local machine, click on Choose file to upload. Alternatively, you can paste the URL for the file into the Or provide file URL: box. Click Run at the bottom of the page. When your job is done, click View reesults.
-
20 variants were analysed, of which 1 is novel.
-
Only 1 gene is affected by variants in this file. The gene has 2 transcripts and both are affected by the variants.
- You can find a list of calculated variant consequences and their impact here.
Yes, the novel variant results in a stop_lost in TraesCS3A02G301400.1 and is a downstream_gene_variant for TraesCS3A02G301400.2.
- Use the filters to view only variants with HIGH impact (you may need to add the column under Show/hide columns at the top of the table if you cannot find it). The filters are found above the detailed results table in the middle. Select Impact and is from the drop-down menus. Then type
HIGHinto the box; this will autocomplete. Click Add.There are 3 variants with high impact and all three are stop altering. Missense variants are not classified as high impact, because they do not always have significant impacts on protein functions. Usually the protein is still produced. In contrast, stop altering variants affect the protein length, and therefore likely affect the protein function.
- At the top right of the table there is an option to download data. Click on VCF for the All option. Open the VCF file you have downloaded in a text editor. You can see that VEP adds annotation in the INFO column of the VCF file.
Web VEP analysis of variants in Coffea canephora (coffee)
You have done whole-genome sequencing and variant-calling experiments for Coffea canephora. You have a VCF file with a small subset of variants from this experiment. Analyse the variants in this file with the VEP tool in Ensembl Plants and determine the following:
1 35246062 var1 C A
1 35246078 var2 G C
1 35246154 var3 T A
-
How many genes and transcripts are affected by variants in this file?
-
Filter the table to find variants with high impact. How many variants have high impact and what consequence predictions do they refer to? Why do you think missense variants are not classified as high impact?
-
Can you export all the results to a VCF file? Compare it to the input VCF file to see what information the VEP adds.
Go to any Ensembl Plants page and click on Tools in the navigation bar at the top of the page. Click on Variant Effect Predictor and change your species to Triticum aestivum by clicking on Change species.
Enter a descriptive name for your VEP job. Paste the variants in VCF format into the text box. Click Run at the bottom of the page. When your job is done, click View reesults.
-
Only 1 gene is affected by variants in this file. The gene has 1 transcript which is affected by the variants.
- Use the filters to view only variants with HIGH impact (you may need to add the column under Show/hide columns at the top of the table if you cannot find it). The filters are found above the detailed results table in the middle. Select Impact and is from the drop-down menus. Then type
HIGHinto the box; this will autocomplete. Click Add.There are 2 variants with high impact (var1 ands var3). One is a start lost (although also a start retained variant), the other is a splice donor variant. Missense variants are not classified as high impact, because they do not always have significant impacts on protein functions. Usually the protein is still produced. In contrast, start/stop altering variants affect the protein length, and therefore likely affect the protein function.
- At the top right of the table there is an option to download data. Click on VCF for the All option. Open the VCF file you have downloaded in a text editor. You can see that VEP adds annotation in the INFO column of the VCF file.
VEP for chicken data
We have identified a few variants associated with body size in chicken (bGalGal1.mat.broiler.GRCg7b):
chr 6, genomic coordinate 23650222, alleles A/C, forward strand
chr 6, genomic coordinate 23645685, alleles C/A, forward strand
chr 1, genomic coordinate 51237121, alleles C/T, forward strand
(a) Which genes and transcripts do these variants map to?
(b) What are the consequence terms for these variants?
(c) Which regulatory feature is affected by the variants?
Go to the Variant Effect Predictor (VEP) under Tools on the top banner of any Ensembl page.
Copy the following into the Paste data text box: 6 23650222 23650222 A/C + var1, 6 23645685 23645685 C/A + var2, 1 51237121 51237121 C/T + var3,
Note that this is the Ensembl default format (chr start end reference/alternate alleles). For additional formats accepted by VEP, have a look here: http://www.ensembl.org/info/docs/tools/vep/vep_formats.html
Click Run.
(a) In the Results table, you’ll see that the variants fall into three genes.
(b) The consequence terms are listed in the Consequence column and Consequences (all) chart and include intron_variant, regulatory_region_variant, upstream_gene_variant and downstream_gene_variant.
(c) Variant 3 at 1:51237121-51237121 with T allele affects regulatory feature ENSR00000006264 (promoter).
VEP for turkey data
My GWAS and sequencing experiments of two groups of turkeys (wild versus domesticated) have identified a few variants associated with body size:
chr 3, genomic coordinate 71541903, alleles C/A, forward strand
chr 18, genomic coordinate 24867, alleles G/A, forward strand
chr 23, genomic coordinate 153938, alleles C/T, forward strand
chr 22, genomic coordinate 10699690, alleles C/G, forward strand
chr 1, genomic coordinate 48884151, alleles A/G, forward strand
(a) Which genes and transcripts do these variants map to?
(b) What are the consequence terms for these variants?
(c) What is the most frequent coding consequence observed in this list?
Go to the Variant Effect Predictor (VEP) under Tools on the top banner of any Ensembl page.
Copy the following into the Paste data text box:
3 71541903 71541903 C/A
18 24867 24867 G/A
23 153938 153938 C/T
22 10699690 10699690 C/G
1 48884151 48884151 A/G
Note that this is the Ensembl default format (chr start end reference/alternate alleles). For additional formats accepted by VEP, have a look here: http://www.ensembl.org/info/docs/tools/vep/vep_formats.html
Click Run.
(a) In the Results table, you’ll see that the variants fall into nine genes (one is intergenic).
(b) The consequence terms are listed in the Consequence column and Consequences (all) chart and include intron_variant, upstream_gene_variant, missense_variant, and downstream_gene_variant.
(c) The most frequent coding consequence is missense_variant. This is shown in one of the pie charts above the table.
VEP analysis of variant in Atlantic salmon
You have performed sequencing and variant-calling experiments for Atlantic salmon. You have a few variants in the VCF format from this experiment:
25 4297825 . G A.
25 4293985 . C G.
25 4294047 . G T.
25 4294047 . G T.
25 4270019 . G A.
(a) How many variants were analysed? How many are novel?
(b) How many genes and transcripts are affected by these variants?
(c) Do any of the variants have different consequences for different transcripts?
(d) Can you export all the results to a VCF file?
Go to www.ensembl.org and click on the Variant Effect Predictor link on the homepage. Click Launch VEP.
Choose Atlantic salmon as the species and enter the five variants from the exercise.
Note: Variation data input can be done in a variety of formats. See more details here http://www.ensembl.org/info/docs/variation/vep/vep_formats.html
Click Run.
When your job is listed as Done, click View Results.
(a) Five variants were analysed, none of these variants are novel.
(b) Only one gene (cdc16) is affected by these variants. It has ten transcripts, all of which are affected.
(c) Yes. These variants have the missense_variant, intron_variant and downstream_gene_variant consequences for the different transcripts of cdc16 gene.
(d) At the top right of the table there is an option to download data. Click on VCF for the All option. Open the VCF file you have downloaded in a text editor. You can see that VEP adds annotation in the INFO column of the VCF file.
Comparative genomics
Demo: gene trees and homology predictions
Plants Compara
Gene trees
Let’s look at the homologues of Triticum aestivum (wheat) TraesCS3D02G007500. Open Ensembl Plants, search for the gene and go to the Gene tab.
Click on Plant compara: Gene tree, which will display the current gene in the context of a phylogenetic tree used to determine orthologues, paralogues and homoeologues.

Funnels indicate collapsed nodes. We can expand them by clicking on the node and selecting Expand this sub-tree from the pop-up menu.
We can also see the protein alignment of the sub-tree by clicking on Wasabi viewer, which will open a pop-up:
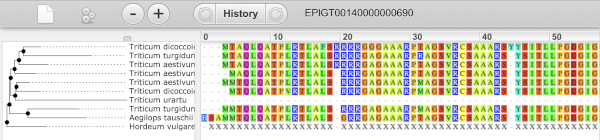
You can download the tree in a variety of formats. Click on the download icon ![]() in the bar at the top of the image to get a pop-up where you can choose your format.
in the bar at the top of the image to get a pop-up where you can choose your format.
Homologues
We can look at homologues in the Orthologues, Paralogues and homoeologues pages, which can be accessed from the left-hand menu. If there are no orthologues, paralogues or homoeologues, then the name will be greyed out. Click on Plant compara: Orthologues to see the orthologues available in plants.
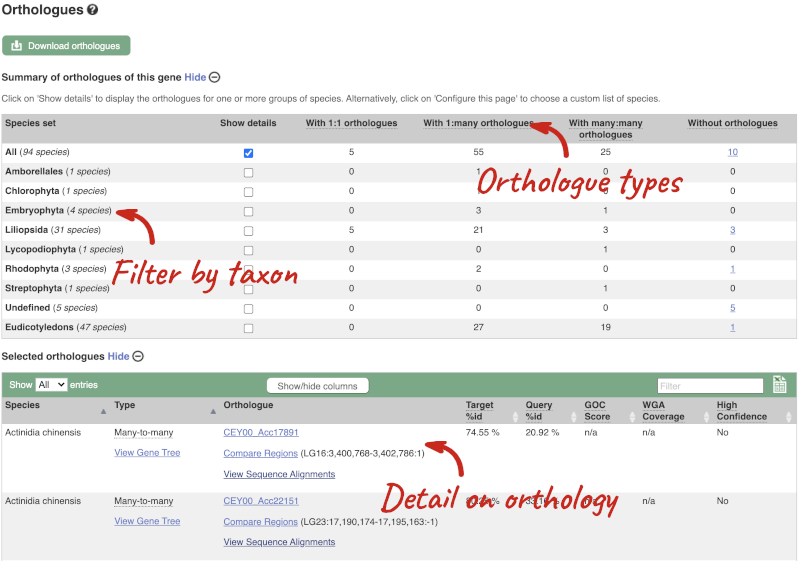
Choose to see only Eudicotyledons orthologues by selecting the box. The table below will now only show details of Eudicotyledons orthologues. Let’s look at Brassica oleracea.
Here we can see there is a many-to-many relationship between the wheat and B. oleracea orthologues. Links from the orthologue allow you to go to alignments of the orthologous proteins and cDNAs. Click on View Sequence Alignments then View Protein Alignment for the first B. oleracea orthologue.
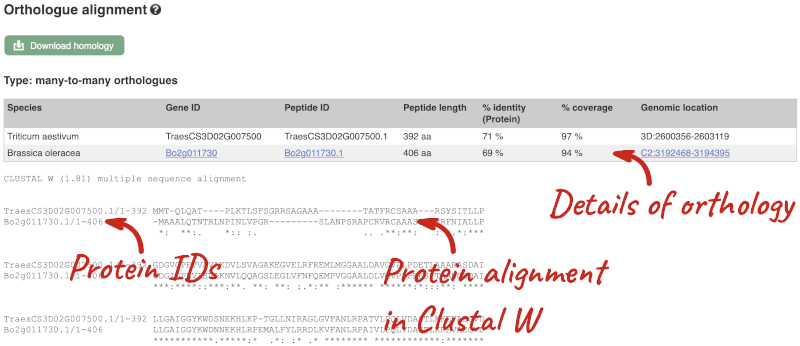
The paralogue page and homoeologue pages are structured in the same way as the orthologue page.
Demo: Whole-genome alignments
Alignments in the Region in Detail view
Let’s look at some of the comparative genomics views in the Location tab. Go to the region 6B:291753000-291966000 in Triticum aestivum (wheat). We can look at individual species comparative genomics tracks in this view by clicking on Configure this page.
Select BLASTz/LASTz alignments from the left-hand menu to choose alignments between closely related species. Turn on the alignments for Triticum dicoccoides (wild emmer), Triticum turgidum (domesticated emmer wheat) and Triticum urartu (red wild einkorn wheat).
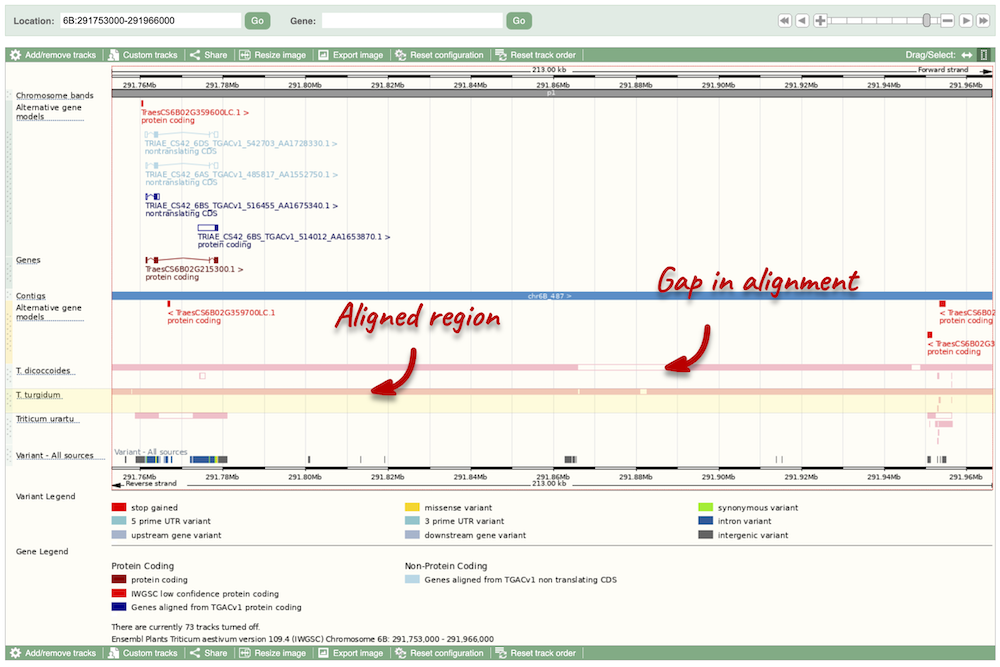
The alignment is greatest between closely related species. We can see that T. turgidum has the most similar sequence to T. aestivum, followed by T. dicoccoides, and T. urartu has the largest gaps in the alignment.
Sequence alignments
We can also look at the alignment between species or groups of species as text. Click on Comparative Genomics: Alignments (text) in the left-hand menu.
Click on Select an alignment to open the alignment menu. Select T. turgidum from the alignments list then click Go.
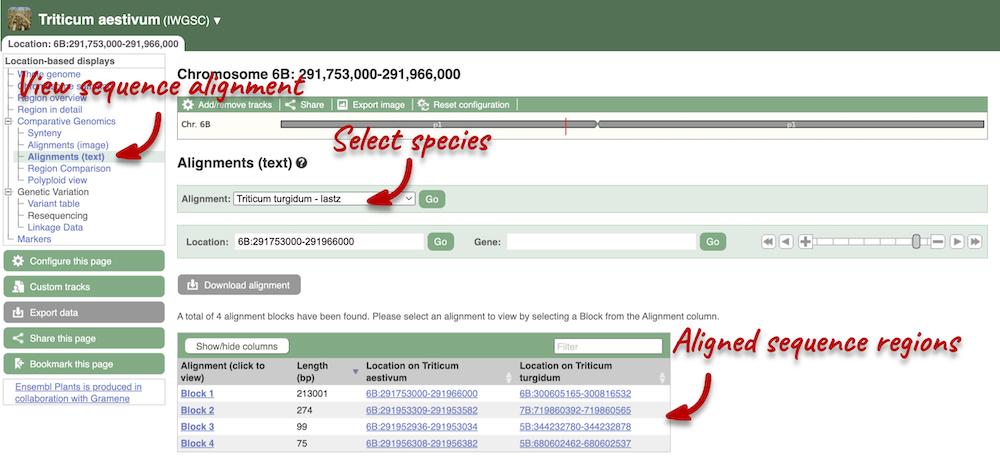
In this case there are 4 blocks aligned of different lengths, some of which correspond to the region we saw unaligned in the image. Click on Block 1.
You will see a list of the regions aligned, followed by the sequence alignment. Click on Display full alignment. Exons are shown in red (you may need to scroll down the page to see the first exon).
Region comparison
To compare with both contigs visually, go to Comparative Genomics: Region Comparison.
To add species to this view, click on the green Select species or regions button. Choose T. turgidum again then close the menu.
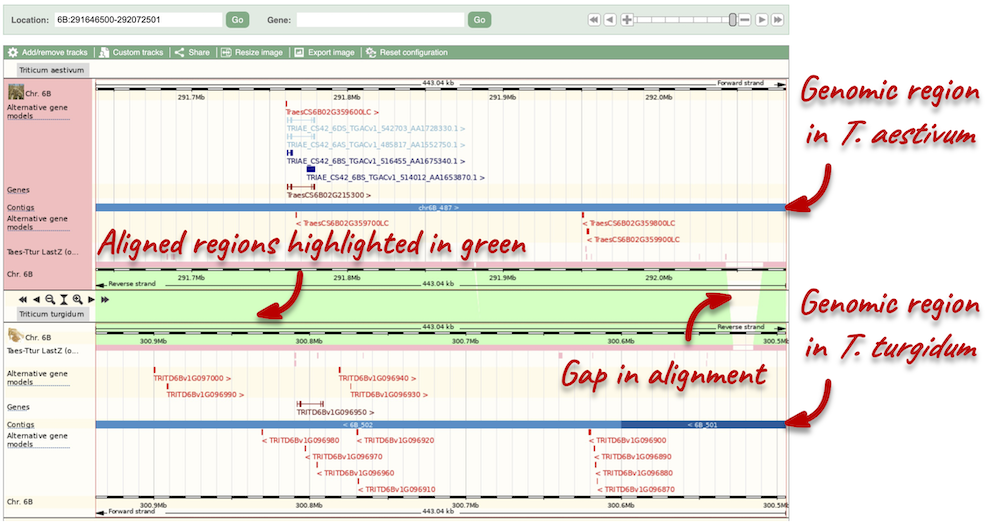
Polyploid view
For polyploids, a Polyploid view will be available for you to compare homologous chromosomes. Genomes for each chromosome are displayed graphically in the lower panel. Your reference chromosome is shown in the first panel. Orange bars show aligned regions between the homologous chromosomes. Aligned regions are also connected and highlighted in green.
Synteny
We can view large-scale syntenic regions from our chromosome of interest. Click on Comparative Genomics: Synteny in the left-hand menu and select T. turgidum* from the **Change species drop-down in the right-hand side.

Black linking lines indicate sequences are oriented in the same directed, red linking lines indicate the sequences are inverted.
Finding orthologous genes for a root transporter in Oryza sativa Japonica (rice)
Search Ensembl Plants for the gene Lsi1 in Oryza sativa Japonica Group (rice). This gene is known to code for an aquaporin transporter that facilitates the uptake of silicon and arsenic through the roots. Silicon concentration is highest in grass species, and is associated with defence.
-
From the gene tab, go to the Orthologues page under Plant Compara. Which plant group has the highest number of 1-to-1 orthologues? Is it the same group that has the highest number of 1-to-many orthologues?
-
Reduce the orthologues table to look only at Triticum aestivum (wheat) orthologues. Why are there three results for a 1-to-1 orthologue?
-
Click on the Compare regions link for chromosome 6B region in wheat to go to the Location tab. Scroll to the bottom image. How do the gene models compare between the species? Do they have the same number of exons?
-
Click back to the Gene tab and click on the Gene gain/loss tree page. Which species has the highest number of members of this gene family? Is it a grass? Can you change the view to see a radial tree?
Go to Ensembl Plants. Look for the main search box highlighted in green. Select Oryza sativa Japonica Group from the drop-down box and type in Lsi1. Click Go and click on the gene ID Os02g0745100.
- Go to Plant Compara: Orthologues on the left-hand panel.
Liliopsida has 24 1-to-1 orthologues, the only group with 1-to-1 orthologues. This group is synonymous with Monocotyledon, so the group that contains the grasses. Eudicotyledons has the highest number of 1-to-many orthologues, indicating that this gene has been duplicated in the eudicots.
- Use the search box in the top right-hand corner of the Selected orthologues table and enter
Triticum aestivum, the table should automatically filter.There are 3 results, one for each component (A,B,D). Note that these are considered 1-to-1 orthologues, rather than 1-to-many. This is because these genes arose in wheat by hybridisation (allopolyploidy), rather than duplication (autopolyploidy).
- Click on Compare regions (found in the 3rd column below the gene identifier) from the 2nd result for component 6B. This takes us to the Location tab. Scroll down to the bottom of the page.
Both genes have 5 exons and the same structure. This looks unusual because the gene in rice is on the forward strand, while the gene in wheat is on the reverse strand. This is reflected in the crossing green links between the pink alignment blocks.
- Click on the Gene tab at the top of the page and click on Gene gain/loss tree in the left-hand panel.
Significant expansions are shown with red branches, and the number of genes in the family shown in the count next to the image and species name. We can see that Echinochloa crus-galli (Cockspur grass) has 25 members in this group.
We can change the tree to radial view by clicking on the icon with two arrows at the top left of the image.
Orthologues, paralogues and gene trees for the maize Zm00001d015746 gene
How many orthologues are predicted for the maize Zm00001d015746 gene in Liliopsida?
How much sequence identity does the Sorghum bicolor protein have to the maize one? Click on the Alignment link next to the Ensembl identifier column to view a protein alignment in Clustal format.
Go to plants.ensembl.org, choose Zea mays and search for Zm00001d015746. Click through to the Gene tab view.
On the gene tab, click on Orthologues at the left side of the page to see all the orthologous genes.
These are the orthologues in the Liliopsida:
- 20 1-to-1
- Seven 1-to-many
- Two many-to-many
The percentage of identical amino acids in the sorghum protein (the orthologue) compared with the gene of interest. i.e. maize Zm00001d015746 (the target species/gene) is 82.07%. This is known as the Target %ID. The identity of the gene of interest (maize GRMZM2G144081) when compared with the orthologue (the sorghum gene, the query species/gene) is 81.69% (the query %ID).
Note the differences in the values of the Target and Query % ID reflects the different protein lengths for the genes.
Homologues and gene trees for the Triticum aestivum (wheat) RHT1 gene
Go to Ensembl Plants and answer the following questions:
-
How many orthologues are predicted for the Triticum aestivum (wheat) gene RHT1 (gene ID TraesCS4D02G040400) gene in Liliopsida?
-
How much sequence identity does the Secale cereale (rye) protein have to the maize one?
-
Download the alignment in Nexus format.
-
Open the gene tree for the wheat RHT1 gene. What is the gene tree ID?
-
How many speciation and duplication nodes does the phylogeny have?
Go to the Ensembl Plants homepage, select Triticum aestivum from the Species drop-down and search for TraesCS4D02G040400. Click through to the Gene tab. On the Gene tab, click on Plant Compara: Orthologues at the left-hand side of the page to see all the orthologous genes.
- These are the orthologues in the Liliopsida:
- 24 1-to-1
- 9 1-to-many
- 0 many-to-many
- Filter the table by entering
Secale cerealein the filter box on the top right-hand corner of the table.The percentage of identical amino acids in the rye protein (the orthologue) compared with the gene of interest (i.e. wheat RHT1; the target species/gene) is 98.71%. This is known as the Target %ID. The identity of the gene of interest (wheat RHT1) when compared with the orthologue (the rye gene, i.e. the query species/gene) is 97.91% (the query %ID).
Note the differences in the values of the Target and Query % ID reflects the different protein lengths for the genes. -
Click on View Sequence Alignments in the Orthologue column. Select View Protein Alignment from the pop-up menu. Click on the green Download homology button above the table and select Nexus. Click on Download or Download Compressed to save the alignment on your local machine.
- Go to Plant Compara: Gene tree in the left-hand menu. You can find the gene tree ID above the phylogeny.
The gene tree ID is EPlGT00940000163877.
- You can find some summary statistics below the gene ID.
There are 418 speciation nodes and 149 duplication nodes.
Exploring whole-genome alignments for Triticum aestivum (wheat)
Go to Ensembl Plants and answer the following questions:
-
Find the TraesCS2D02G080000 gene in Triticum aestivum (wheat). What is the function for this gene and what are its coordinates?
-
Go to the Location tab. Turn on the LASTZ-net alignment tracks for Arabidopsis thaliana, Zea mays (corn) and Sorghum bicolor (great millet). Are there any regions where you can see gaps in in some of the species alignments?
-
Go to the Region comparison view and compare to A. thaliana. What occurs at this gap in the alignment?
-
Export the Block 2 alignment between T. aestivum and A. thaliana in ClustalW format.
- Go to the Ensembl Plants homepage. Select Triticum aestivum from the Species drop-down, enter
TraesCS2D02G080000in the search box and click Go. Open the Gene tab.The gene description is as follows: Ascorbate peroxidase, ROS homeostasis, Chloroplast protection, Carbohydrate metabolism, Plant architecture, Fertility maintenance. This was projected from Oryza sativa (Os07g0694700).
- Go the Location tab in the top left-hand corner. Click on CConfigure this page in the side menu. Open Comparative genomics: BLASTz/LASTz alignments in the pop-up menu. Turn on the tracks for Arabidopsis thaliana, Zea mays (corn) and Sorghum bicolor (great millet) in the Normal style. Save and close the pop-up menu
There is alignment across most of the coding regions, with some gaps occurring in all 3 species. These gaps map with the intronic regions of the T. aestivum gene.
- Click on Comparative Genomics: Region Comparison in the left-hand menu. Go to the Select species or regions button and add A. thaliana. Save and close the menu.
The gap in the alignment translates to the intronic regions of the T. aestivum gene.
- Go to Comparative Genomics: Alignments (text) and select A. thaliana from the Alignment drop-down. Click on the green Download alignment button and select ClustalW. Download the file to your local machine either in a compressed format, or as it is by clicking the green Download button above the file format preview.
Finding orthologous genes for disease resistance gene in Coffea canephora (coffee)
Resistance to the leaf rust delivered by SH3 factor(s) is well-grounded as specially durable. in 2023, Paula Cristina da Silva Angelo et al (https://doi.org/10.1016/j.pmpp.2023.102111) reported that the Arabidopsis thaliana gene AT1G50180 is an important gene in the SH3 locus conferring diseae resistance.
Search Ensembl Plants for the gene AT1G50180 in Arabidopsis thaliana.
-
From the gene tab, go to the Arabidopsis thaliana AT1G50180 gene Orthologues page under Plant Compara.
-
Reduce the orthologues table to look only at Coffea canephora (coffee) orthologues. How many results can you see?
-
Download the cDNA alignment in ClustalW format for the alignment between the Arabidopsis thaliana AT1G50180 gene and the Coffea canephora GSCOC_T00030728001 gene.
Go to Ensembl Plants. Select Arabidopsis thaliana from the drop-down box and type in AT1G50180. Click Go and click on the gene ID AT1G50180.
-
Go to Plant Compara: Orthologues on the left-hand panel.
- Filter for Coffea canephora using the filter option in the top right hand corner of the table.
Coffee has 25 many-to-many orthologues.
- Click on View Sequence Alignments then cDNA (found in the 3rd column below the gene identifier) for the GSCOC_T00030728001 gene. This takes us to the Orthologue Alignment page.
Click on Download Homology to download the alignment in ClustalW format
Orthologues and gene trees for the Gallus gallus (Chicken) BRAF gene
- Let’s explore the orthologues of the chicken BRAF gene.
- How many orthologues are predicted for the chicken BRAF in sauropsida (birds and reptiles)?
- How much sequence identity does the Anolis carolinensis (Green anole) protein have to the chicken one?
- Export the protein alignment in Clustal format.
- Look at the orthologue in human. Is there a genomic alignment between human and chicken? Is there a gene for both species in this region?
- Go to the Ensembl homepage, select Chicken from the drop-down list in the blue search box, enter BRAF and click Go. Open the **Gene tab and click on Comparative Genomics: Orthologues at the left-hand panel to see all the orthologous genes.
There 25x 1:1 and 1x 1:many orthologues in sauropsida.
Find Green anole in the Selected orthologues (you can use the filter in the top right-hand corner).
The percentage of identical amino acids in the Green anole protein (the orthologue) compared with the gene of interest. i.e. chicken BRAF (the target species/gene) is 84.42%. This is known as the Target %id. The identity of the gene of interest (chicken BRAF) when compared with the orthologue (Green anole BRAF, the query species/gene) is 94.67% (this is the Query %id).
Click on the View Sequence Alignments link in the Orthologue column of the Selected orthologues table and select View Protein Alignment in the pop-up menu. To download the alignment, click on the Download homology button and select the CLUSTALW file format in the pop-up menu.
- Click on Comparative Genomics: Genomic alignments in the left-hand panel. Click on Select an alignment and add Human in the pop-up menu. In the table, select Block 1 to view the largest block of aligned sequence (this will lead you to the Location tab). Click on Display full alignment. In the alignment, sequences coloured in red are exons.
There is a gene in both species in this region. You can find where the start and stop codons are located if you Configure this page and select Codons: START/STOP codons in the options.
Note: You can visualise the alignment in the genomic context in the Comparative Genomics: Region Comparison page (blue lines connect homologous genes between species). Go to Select species or regions, add Human and close the pop-up menu. Click on Configure this page. In the pop-up menu under Comparative features category, enable the Join genes option. You may need to zoom out on the Region in detail view to see blue lines connecting all the homologous genes between chicken and human genes in that region.
Whole-genome alignments in Sus scrofa (Pig)
Go to www.ensembl.org to find the DBH gene on the reference pig genome (Sscrofa11.1).
-
Go to the Location page for this gene. View the Alignments (image) and Alignments (text) for the 16 pig breeds EPO-Extended. Do all the pig breeds show a gene in these alignments?
-
Export the alignments in ClustalW format.
-
Go to the Region in detail view and turn on the 16 pig breeds EPO-Extended multiple alignment, conservation score and constrained elements tracks. Are there any differences between the conservation score and constrained elements tracks?
-
Compare the 16 way GERP elements track and the 91 way GERP elements track that is already turned on by default.
- What is the difference between the two tracks?
- Which regions of the gene do most of the constrained element blocks match-up to?
- How can you find more information on how the constrained elements track was generated?
- Search for the DBH(ENSSSCG00000005742) gene in the Pig (SScrofa11.1) reference and switch to the Location tab. Click on Alignments (image) in the left-hand panel. Under Alignment, click on the Select alignment button to open a pop-up menu. Enable the 16 pig breeds EPO-Extended alignment, then close the menu.
All 12 big breeds as well as cow, horse and sheep have an alignment at this region. This can also be seen in the Alignments (text) page, where the exons are coloured in red.
-
You can export the alignments from either the Alignments (images) or Alignments (text) pages. Click on the blue Download alignments button at the top of the page. From the pop-up menu, select File format: CLUSTALW. You can Preview the alignment in a new browser tab, or Download the file to your local machine.
- Click on Region in detail in the left-hand panel. In the pop-up menu, go to the Comparative genomics section and turn on the following tracks:
- Multiple alignments: 16 pig breeds EPO-Extended
- Conservation score for 16 pig breeds EPO-Extended
- Constrained elements for 16 pig breeds EPO-Extended
Close the pop-up menu and find the tracks in the Region in detail view.
The 16 pig breeds EPO-extended track shows that the entire region for the DBH gene can be aligned among the big breeds and related agricultural species. The Constrained elements and Conservation score tracks show where the conserved sequence is located in the alignment. Regions where constrained elements are found are regions with high GERP scores. Higher conservation regions (i.e. constrained elements) match up with exonic regions (exons tend to be highly conserved) of the gene. Note that there are intronic regions that seem to be fairly conserved across the species available.
- For both 16 way GERP elements and 91 way GERP elements tracks, click on the track name to open the pop-up menu. Hover over the i icon with your cursor to find a track description.
The 16 way GERP elements track shows the 16 pig breeds EPO-Extended multiple whole-genome alignment. The 91 way GERP elements track shows the 91 eutherian mammals EPO-Extended multiple whole-genome alignment.
You can move the 91 way GERP elements track closer to the 16 way GERP elements track to make any comparisons easier.
You will notice that constrained elements match-up with exonic regions in the genome.
Click on the track name and open the information tab in the pop-up menu. Click on the GERP conservation score link.
This opens the documentation page for the multiple whole-genome alignment calculations.
Gene trees and homologues in Sus scrofa (Pig) breeds
We are going to look at the PLAG1 gene in the pig reference (Sscrofa11.1) genome and explore its gene tree and homologues.
-
Have orthologues been identified in any pig breeds? If so, which ones?
-
Open the cDNA sequence alignment against the Tibetan breed. What does the asterisk (*) symbol mean in the alignment? What is the % identity (cDNA) and what does the number stand for?
-
Let’s look at the PLAG1 breed gene tree. How many genes are depicted in the gene tree? According to the gene tree, which orthologue is the most closely related to the pig reference PLAG1 gene? What is the Ensembl ID of the orthologue and what strand is it located on?
-
Export the gene tree in Newick format.
- Open the Gene tab for the PLAG1 gene (ENSSSCG00000006247) in the pig reference (Sscrofa11.1) genome. In the left-hand panel, click on Comparative Genomics: Breeds: Orthologues to explore orthologues in pig breeds.
PLAG1 orthologues have been identified in all 12 available pig breeds: Bamei, Berkshire, Hampshire, Jinhua, Large white, Meishan, Pietrain, Rongchang, Tibetan, Wuzhishan and USMARC.
- Stay in the Comparative Genomics: Breeds: Orthologues page and look for the Pig - Tibetan entry in the Orthologues table. Click on View Sequence Alignments and select View cDNA Alignment in the pop-up menu. This will lead you to the sequence alignment between the pig reference PLAG1 gene and the orthologue in the Tibetan breed. Click on the ? icon above the alignment to open the corresponding help page and find a description of the data (including descriptions of the conservation codes and summary statistics).
The alignment is in ClustalW format and the asterisk (*) is a conservation code that denotes the aligned nucleotides are identical in both sequences.
In the **Type: 1-to-1 orthologues table, look for the column **% identity (cDNA).
The % identity (cDNA) describes the number of identical nucleotides between the two sequences in the alignment. The % identity (cDNA) between the pig reference and the Tibetan breed is 99%, meaning 99% of the cDNA sequence alignment is identical.
- In the left-hand panel, click on Comparative Genomics: Breeds: Gene tree. Above the gene tree image, you can find some summary statistics.
There are 16 genes in the gene tree.
In the gene tree, look for PLAG1, Pig (coloured in red) in the tree and find the closest branch.
The most closely related orthologue is the PLAG1 gene in the Berkshire breed. Clicking on PLAG1, Pig - Berkshire opens more details on the orthologue including the Ensembl ID (ENSSSCG00065079212) and strand (reverse).
- Click on the Export button at the top of the gene tree image. In the pop-up menu, select File format: Newick (you can find a preview of each file format underneath) and Preview the file on your browser.
Exploring European seabass pax6b orthologues
Go to Ensembl to answer the following questions:
-
How many orthologues are predicted for the Dicentrarchus labrax (European seabass) pax6b gene across ray-finned fishes?
-
How much sequence identity does the Sparus aurata (Gilthead seabream) protein have to the European seabass one?
-
Can you tell which end of the pax6b protein is more conserved between these two species by looking at the orthologue alignment?
- From the Ensembl homepage, choose European seabass from the drop-down list and search for
pax6b. Click through to the Gene tab view. Click on Comparative Genomics: Orthologues at the left side of the page to see all the orthologous genes.From the summary table, we can see that the European seabass has 52 1-to-1 orthologues and 9 1-to-many orthologues in the ray-finned fishes species list.
- Search for
Gilthead seabreamin the orthologues table below.The percentage of identical amino acids in the Gilthead seabream protein (the orthologue) compared with the gene of interest, i.e. the European seabass pax6b (the target species/gene), is 99.78%. This is known as the Target%id.
The identity of the gene of interest (European seabass pax6b) when compared with the orthologue (Gilthead seabream pax6b, the query species/gene) is 99.78% (the Query %id).
Note that any difference in the values of the Target and Query %id reflects the different protein lengths for the two orthologues. In the case of the European seabass and Gilthead seabream, the identity is the same as both proteins are 457 amino acids long.
- Click on the View Sequence Alignments link in the Orthologue column to View Protein Alignment in ClustalW format.
Conserved amino acids are indicated by asteriks (*). You can find more information about the ClustalW format in our help pages. There is no difference between the N-terminus and C-terminus. The sequence is conserved around both termini.
BioMart
Demo: BioMart
Follow these instructions to guide you through BioMart to answer the following query:
- What genes are found on chromosome 5D, between 19400000 and 21300000 in wheat?
- What are the NCBI Gene IDs for these genes?
- Are there associated functions from the GO (gene ontology) project that might help describe their function?
- What are their cDNA sequences?
Step 1: Choose the database and dataset
Click on BioMart in the top header of any Ensembl Plants page to open BioMart
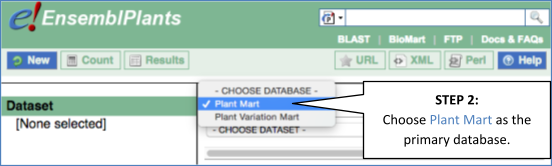
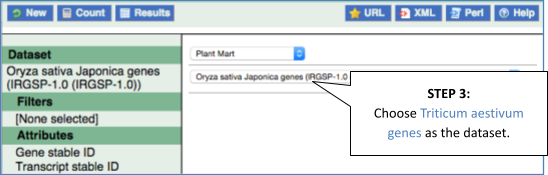
Step 2: Choose appropriate filters
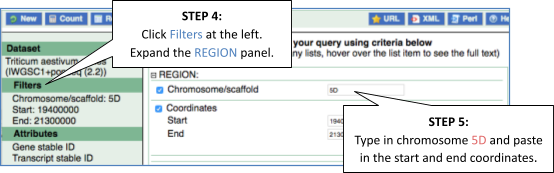
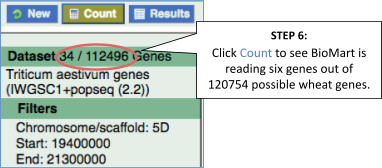
Step 3.1: Select attributes (features)
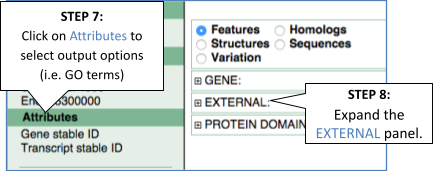


Step 4.1: Get the results
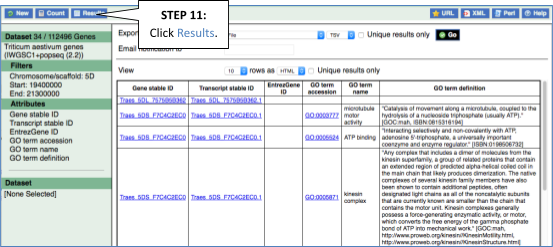
Why are there multiple rows for one gene ID? For example, look at the first few rows.
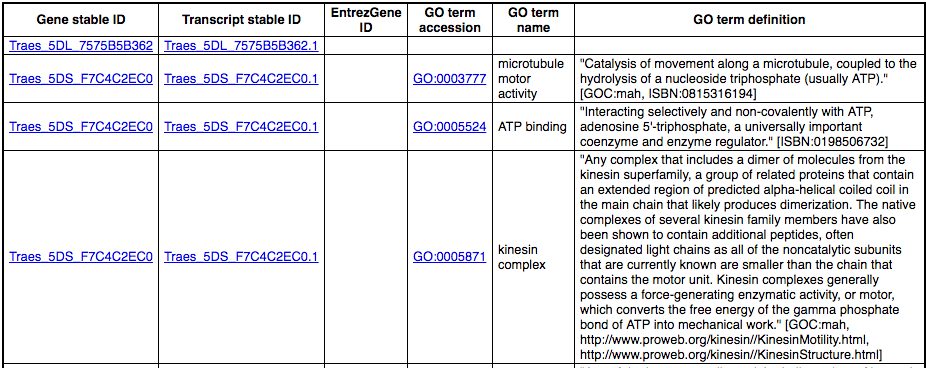
Step 3.2: Select attributes (sequences)
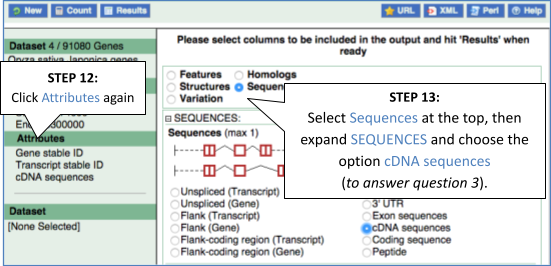
Step 4.2: Get the results
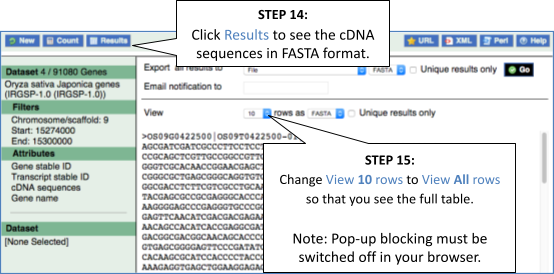
Note: you can use the Go button to export a file.
What did you learn about the wheat genes in this exercise?
Could you learn these things from the Ensembl browser? Would it take longer?
For more details on BioMart, have a look at this publication: Kinsella RJ, Kähäri A, Haider S, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database : the Journal of Biological Databases and Curation. 2011 ;2011:bar030. DOI: 10.1093/database/bar030. PMID: 21785142; PMCID: PMC3170168.
BioMart Convert IDs
BioMart is a very handy tool when you want to convert IDs from different databases. The following is a list of 20 IDs of wheat proteins from the NCBI RefSeq database:
NP_114254.1 NP_114277.1 NP_114275.1 NP_114283.1 YP_398395.1 NP_114279.1 NP_114274.1 NP_114273.1 NP_114273.1 NP_114265.1 NP_114247.1 NP_114243.1 NP_114276.1 NP_114276.1 NP_114262.1 NP_114287.1 NP_114239.1 NP_114276.1 NP_114243.1 NP_114280.1
Generate a list that shows to which Ensembl Gene IDs and to which gene names these RefSeq IDs correspond. Do these 20 transcripts correspond to 20 genes?
Click New. Choose the Ensembl Plants Genes database. Choose the Triticum aestivum genes dataset.
Click on Filters in the left panel. Expand the GENE section by clicking on the + box. Select Input external references ID list - RefSeq peptide ID(s) and enter the list of IDs in the text box (either comma separated or as a list). HINT: You may have to scroll down the menu to see these.
Count shows 68 genes (the hybridisations and whole genome duplications in wheat’s evolutionary history means that many RefSeqs are duplicated across the genome)..
Click on Attributes in the left panel. Select the Features attributes page. Expand the Gene section by clicking on the + box. Select Gene name from the Gene section. Expand the External section by clicking on the + box. Select RefSeq Peptide ID from the External References section.
Click the Results button on the toolbar. Select View All rows as HTML or export all results to a file.
Get genes by protein domain
Go to Ensembl Plants and find the following information:
Retrieve the protein sequences (in FASTA format) of all Triticum aestivum (wheat) genes that have an NCBI Gene ID, that are protein-coding and with Transmembrane helices. Do a count after the selection of each filter to check the number of genes remaining in your dataset. Export the results of the sequences and select Gene description and Source of gene name as headers.
-
Click on BioMart on the navigation bar at the top of the page. Click the New button on the toolbar on the top left-hand corner, choose the Ensembl Plants Genes database and Triticum aestivum genes (IWGSC) dataset. Now, filter for the genes with NCBI Gene ID only:
- Click on Filters in the left panel, expand the GENE section by clicking on the + box. Select with NCBI Gene ID under Limit to genes (external references)…. Make sure the box next to the filter is ticked, otherwise the filter won’t work. Click the Count button on the toolbar.
This will give you 92 Genes.
Now filter further for genes that are protein-coding by selecting Gene type – protein_coding and click again on Count.
This still gives you 92 Genes, meaning that all genes you have previously filtered are protein-coding.
Finally, filter for genes that have a signal peptide domains. Expand the PROTEIN DOMAINS AND FAMILIES section by clicking on the + box. Select Transmembrane helices – Only under Limit to genes….
There are 79 genes on the bread wheat genome that contain NCBI Gene IDs and protein coding with signal domains.
-
Go to Attributes on the left-hand panel. Select Sequences from the options on the right. Expand the SEQUENCES section by clicking on the + box and select Peptide. Select the appropriate header information from the HEADER INFORMATION section: Gene description and Source of gene name.
- Click on Results on the toolbar and the sequence will be shown as FASTA format. You can export the sequence by downloading it directly to your local machine or sending it to your email.
Export homologues
For a list of Hordeum vulgare genes, export the Triticum aestivum orthologues.
HORVU.MOREX.r3.2HG0191020
HORVU.MOREX.r3.2HG0134140 HORVU.MOREX.r3.2HG0144470 HORVU.MOREX.r3.1HG0064460 HORVU.MOREX.r3.1HG0008890
Go to plants.ensembl.org and click on the link Tools at the top of the page. Click on BioMart.
Click New. Choose the Ensembl Plants Genes database. Choose the Hordeum vulgare genes (MorexV3_pseudomolecules_assembly) dataset.
Click on Filters in the left panel. Expand the GENE section by clicking on the + box. Enter the gene list in the Input external references ID list box. Select Gene stable ID(s) [e.g. HORVU.MOREX.r3.1HG0000030] from the drop-down menu.
Click on Attributes in the left panel. Select the Homologues attributes page. Expand the ORTHOLOGUES section by clicking on the + box. Select Triticum aestivum gene stable ID.
Click Results.
Retrieve a list of SNPs from the tomato genome
The region between coordinates 21,394,819 and 21,397,868 on chromosome 6 in tomato contains a gene involved in oxidation-reduction process (GO:0055114).
Can I use BioMart to retrieve all the SNPs in this region including their IDs and possible alleles?
Click the New button on the toolbar, choose the Ensembl Plants Genes database and Solanum lycopersicum genes (SL2.40 (ITAG2.3)) dataset.
Click on Filters in the left panel and expand the REGION section. Under Multiple regions enter 6:21394819-21397868.
Click on Attributes in the left panel, select the Variation attribute and under VARIANT ASSOCIATED INFORMATION, tick Variant_name and Variant alleles.
Exporting homologues with BioMart
Go to Ensembl Plants’s BioMart. For a list of Arabidopsis thaliana genes, export the coffee orthologues:
MLP28, MEE18, EP1, QRT3, MOT2, GC4, WYR
Do all of these genes have a homologue in coffee?
-
Go to BioMart (you can find a shortcut in the navigation bar at the top of any Ensembl Plants page) and click New. Choose the Ensembl Plant Genes database. Choose the Arabidopsis thaliana genes (TAIR10) dataset.
-
Click on Filters in the left panel. Expand the GENE. Enter the gene list in the Input external references ID list box. Select Gene Name from the input options dropdown list.
-
Click on Attributes in the left panel. Select the Homologues attributes at the top of the page. Expand the GENE section. Select Gene Name. Expand the ORTHOLOGUES [A-E] section. Select Coffea canephora gene stable ID.
-
Click Results. Select View: All rows as HTML.
All genes have a homologue in coffee.
Export sequences in FASTA format
Retrieve the sequences of all chicken genes (Gallus gallus) that are located on chromosome 20, that are protein coding and that encode for proteins containing transmembrane domains. Do a count after selection of each filter to check the number of genes remaining in your dataset. Export the results of the protein sequences (FASTA) as Compressed web file and get the results notified to you by email.
On the Ensembl homepage, click on the BioMart link on the toolbar.
Start with all genes in chicken by choosing the Ensembl Genes database, then Gallus gallus genes dataset.
Now, filter for the genes on the 20 chromosome only:
Click on Filters in the left panel, expand the REGION section by clicking on the + box. Select Chromosome – 20.
Now click the Count button on the toolbar.
This will give you 473 / 24356 Genes.
Now filter further for genes that are protein-coding by expanding the GENE section (simply click on the + box). Then select Gene type – protein_coding and click again on Count.
This now gives you 332 / 24356 Genes.
Finally, filter for genes that encode proteins that contain transmembrane domains. Expand the PROTEIN DOMAINS section by clicking on the + box. Select Transmembrane helices – Only.
There are 69 genes on chromosome 20 in chicken that are protein coding and contain transmembrane domains.
Now you can specify the attributes to be included in the output (note that a number of attributes will already be selected by default). Click on Sequences, then Protein. The sequence will be exported as FASTA format.
Have a look at a preview of the results (only 10 rows of the results will be shown):
Click the Results button on the toolbar.
If you are happy with how the results look in the preview, output all the results by selecting Export all results to, then choose the Compressed web file (notify by email), click on _Unique results only, enter your email address in the appropriate box and click on Go.
BioMart: Finding genes by protein domain
Find chicken proteins with transmembrane domains located on chromosome 9.
As with all BioMart queries you must select the dataset, set your filters (input) and define your attributes (desired output). For this exercise:
Dataset: Ensembl genes in chicken
Filters: Transmembrane proteins on chromosome 9
Attributes: Ensembl gene and transcript IDs and Associated gene names
Go to the Ensembl homepage (https://www.ensembl.org) and click on BioMart at the top of the page. Select Ensembl genes as your database and Chicken genes (bGalGal1.mat.broiler.GRCg7b) as the dataset. Click on Filters on the left of the screen and expand REGION. Change the chromosome to 9. Now expand PROTEIN DOMAINS AND FAMILIES, also under filters, and select Limit to genes …, choosing With Transmembrane helices from the drop-down and select Only. Clicking on Count should reveal that you have filtered the dataset down to 143 genes.
Click on Attributes. Under Features expand GENE. Select Gene name.
Now click on Results. The first 10 results are displayed by default; display all results by selecting All from the drop-down menu above the table.
The output will display the Ensembl gene ID, Ensembl Transcript ID and associated gene names of all proteins with a transmembrane domain on chicken chromosome 9. If you prefer, you can also export as an Excel sheet by using the Export all results to XLS option.
BioMart: Find genes associated with array probes
Here are two affymetrix probeset IDs from my microarray experiment that seem to map uniquely to genes in the chicken genome: Gga.12669.1.S1_at, GgaAffx.7784.1.S1_at
(a) Retrieve for the genes corresponding to these probe-sets the Ensembl Gene and Transcript IDs as well as their gene symbols and descriptions.
(b) In order to analyse these genes for possible promoter/enhancer elements, retrieve the 2000 bp upstream of the transcripts of these genes.
(c) In order to be able to study these chicken genes in duck, identify their duck orthologues. Also retrieve the genomic coordinates of these orthologues.
(a) Click New. Choose the Ensembl Genes database. Choose the Chicken genes dataset.
Click on Filters in the left panel. Expand the GENE section by clicking on the + box. Select Input microarray probes/probesets ID list - AFFY Chicken probe ID(s) and enter the list of probeset IDs in the text box (either comma separated or as a list).
Count shows three genes match this list of probesets.
Click on Attributes in the left panel. Select the Features attributes page. Expand the GENE section by clicking on the + box. In addition to the default selected attributes, select Gene name and Gene description. Expand the EXTERNAL section by clicking on the + box. Select AFFY Chicken probe from the Microarray probes/probesets section.
Click the Results button on the toolbar. Select View All rows as HTML or export all results to a file. Tick the box Unique results only.
Your results should show that the 2 probes map to 2 Ensembl genes.
(b) Don’t change Dataset and Filters – simply click on Attributes.
Select the Sequences category. Expand the SEQUENCES tab by clicking on the + box. Select Flank (Transcript) and enter 2000 in the Upstream flank text box. Expand the HEADER INFORMATION tab by clicking on the + box. Select Gene description and Gene name in addition to the default selected attributes.
Note: Flank (Transcript) will give the flanks for all transcripts of a gene with multiple transcripts. Flank (Gene) will give the flanks for one possible transcript in a gene (the most 5’ coordinates for upstream flanking).
Click the Results button on the toolbar.
(c) You can leave the Dataset and Filters the same, and go directly to the Attributes section:
Click on Attributes in the left panel. Select the Homologues category. Expand the GENE tab by clicking on the + box. Select Gene name. Unselect Transcript stable ID and Transcript stable ID version. Expand the ORTHOLOGUES [A-E] tab by clicking on the + box. Select Duck gene stable ID, Duck chromosomes/scaffold name, Duck chromosome/scaffold start (bp) and Duck chromosome/scaffold end (bp).
Click the Results button on the toolbar. Select View All rows as HTML or export all results to a file.
Your results should show that for each chicken gene, one duck orthologue has been identified.