Filter Events by Year
UCL MSc: Navigating the Ensembl Genome Browser
Course Details
- Lead Trainer
- Aleena Mushtaq
- Event Date
- 2023-02-22
- Location
- University College London, UK
- Description
- Work with the Ensembl Outreach team to get to grips with the Ensembl browser.
- Survey
- UCL MSc: Navigating the Ensembl Genome Browser Feedback Survey
Demos and exercises
Hands on
Ensembl Homepage
The front page of Ensembl is found at ensembl.org. It contains lots of information and links to help you navigate Ensembl:
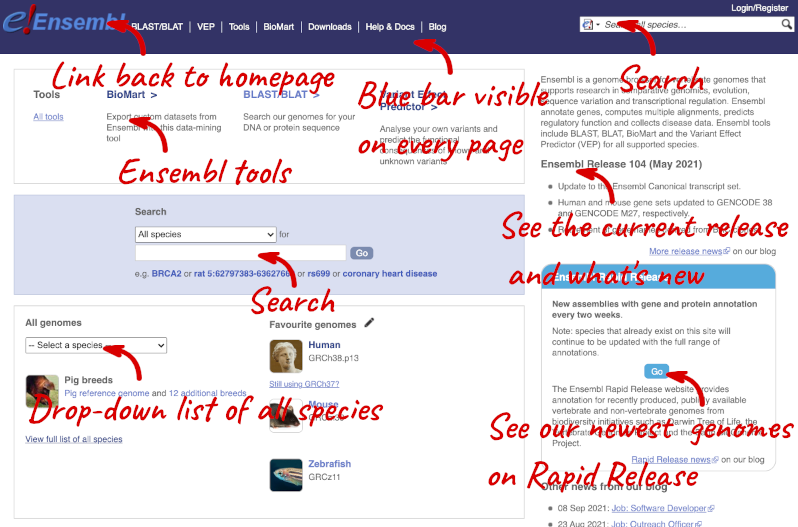
At the top left you can see the current release number and what has come out in this release. To access old releases, scroll to the bottom of the page and click on View in archive site.
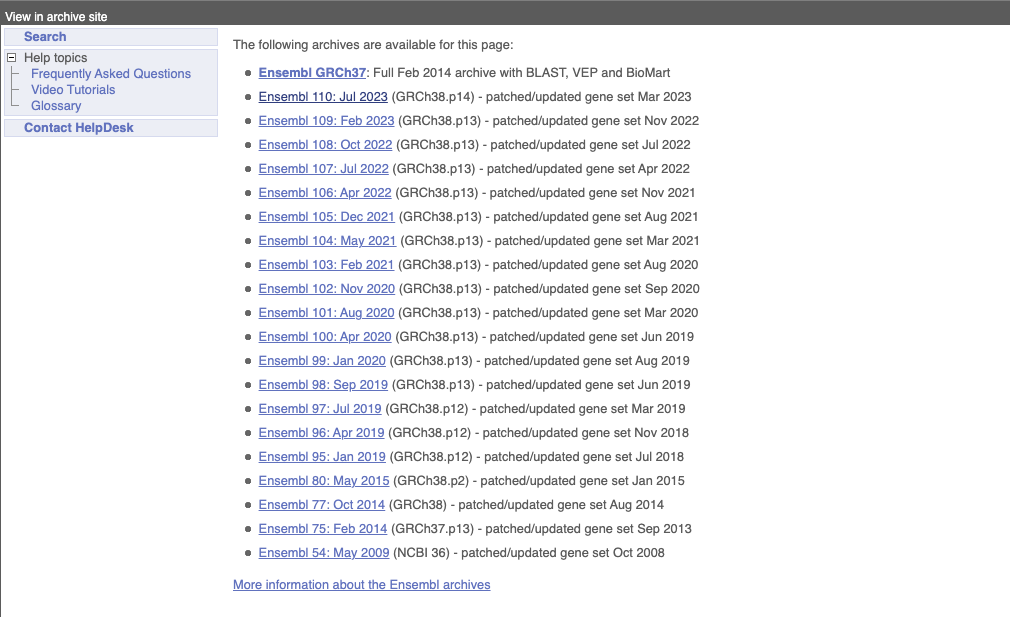
Click on the links to go to the archives. Alternatively, you can jump quickly to the correct release by putting it into the URL, for example e98.ensembl.org jumps to release 98.
Click on View full list of all species.
Click on the common name of your species of interest to go to the species homepage. We’ll click on Human.
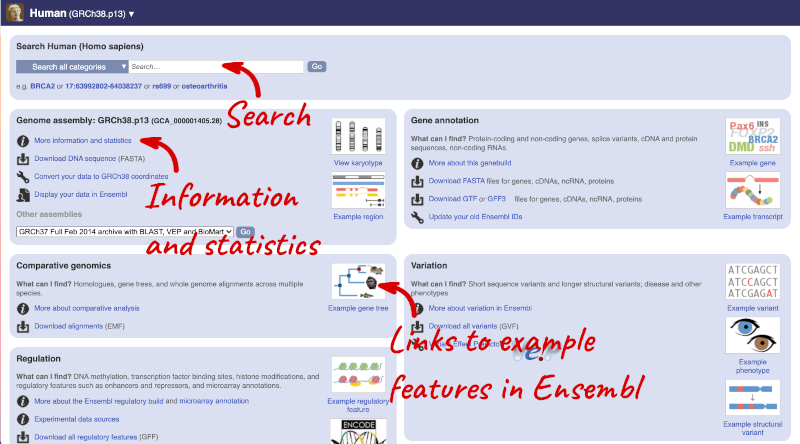
Here you can see links to example pages and to download flatfiles. To find out more about the genome assembly and genebuild, click on More information and statistics.
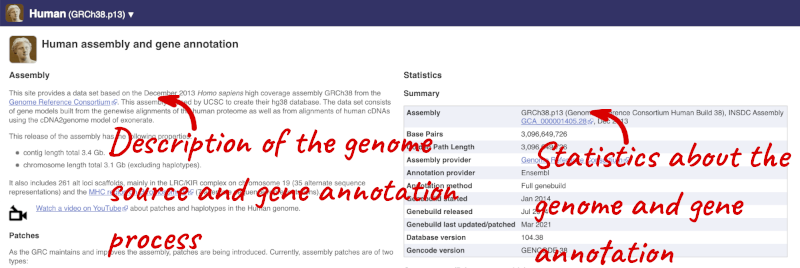
Here you’ll find a detailed description of how to the genome was produced and links to the original source. You will also see details of how the genes were annotated.
The current genome assembly for human is GRCh38. If you want to see the previous assembly, GRCh37, visit our dedicated site grch37.ensembl.org.
Region in detail view
Start at the Ensembl front page, ensembl.org. You can search for a region by typing it into a search box, but you have to specify the species.
To bypass the text search, you need to input your region coordinates in the correct format, which is chromosome, colon, start coordinate, dash, end coordinate, with no spaces for example: human 4:122868000-122946000. Type (or copy and paste) these coordinates into either search box.
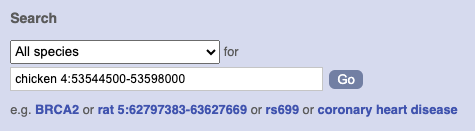 or
or 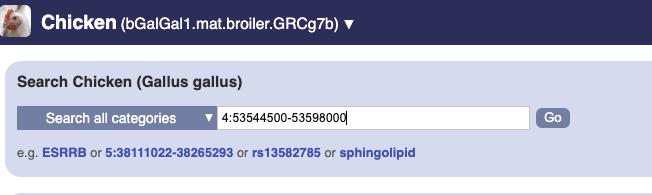
Press Enter or click Go to jump directly to the Region in detail Page.
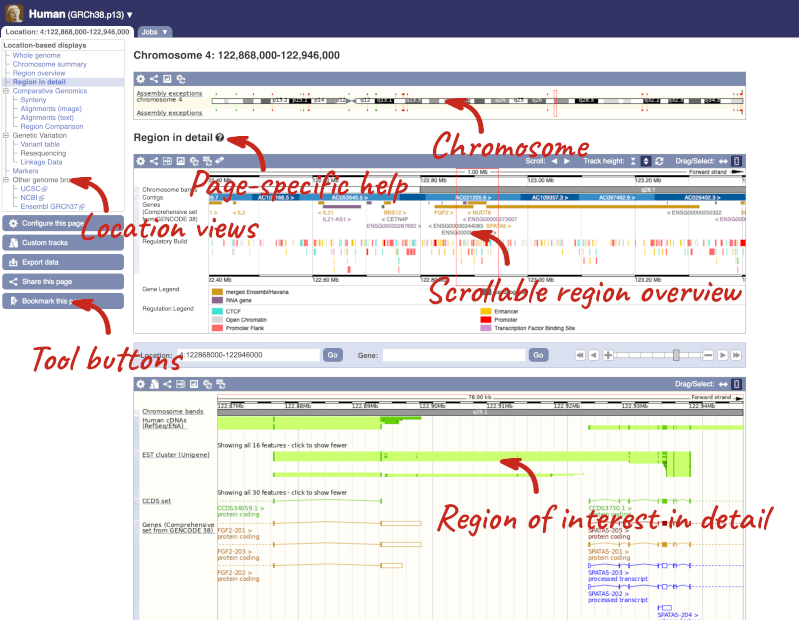
Click on the  button to view page-specific help. The help pages provide text, labelled images and, in some cases, help videos to describe what you can see on the page and how to interact with it.
button to view page-specific help. The help pages provide text, labelled images and, in some cases, help videos to describe what you can see on the page and how to interact with it.
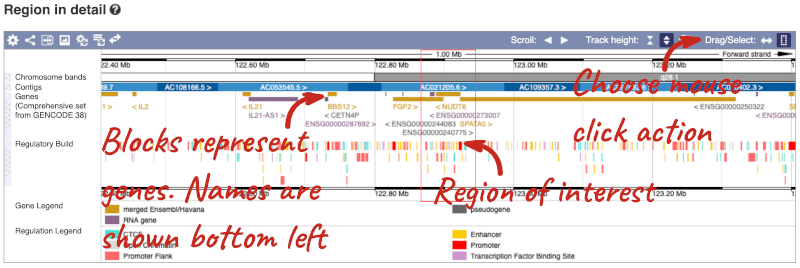
The Region in detail page is made up of three images, let’s look at each one in detail.
- The first image shows the chromosome:
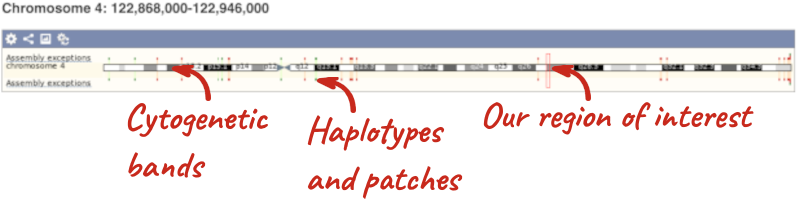
The region we’re looking at is highlighted on the chromosome. You can jump to a different region by dragging out a box in this image. Drag out a box on the chromosome, a pop-up menu will appear.
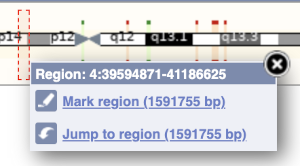
If you wanted to move to the region, you could click on Jump to region (### bp). If you wanted to highlight it, click on Mark region (###bp). For now, we’ll close the pop-up by clicking on the X on the corner.
- The second image shows a 1Mb region around our selected region. This is always 1Mb in human, but the fixed size of this view varies between species. This view allows you to scroll back and forth along the chromosome.
You can also drag out and jump to or mark a region.
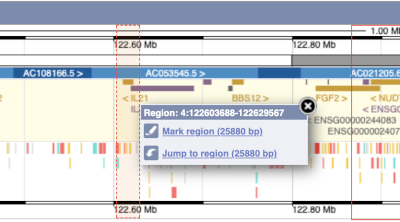
Click on the X to close the pop-up menu.
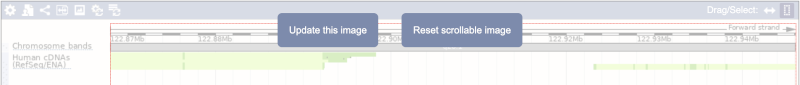
Click on the Drag/Select button  to change the action of your mouse click. Now you can scroll along the chromosome by clicking and dragging within the image. As you do this you’ll see the image below grey out and two blue buttons appear. Clicking on Update this image would jump the lower image to the region central to the scrollable image. We want to go back to where we started, so we’ll click on Reset scrollable image.
to change the action of your mouse click. Now you can scroll along the chromosome by clicking and dragging within the image. As you do this you’ll see the image below grey out and two blue buttons appear. Clicking on Update this image would jump the lower image to the region central to the scrollable image. We want to go back to where we started, so we’ll click on Reset scrollable image.
- The third image is a detailed, configurable view of the region.

Here you can see various tracks, which is what we call a data type that you can plot against the genome. Some tracks, such as the transcripts, can be on the forward or reverse strand. Forward stranded features are shown above the blue contig track that runs across the middle of the image, with reverse stranded features below the contig. Other tracks, such as variants, regulatory features or conserved regions, refer to both strands of the genome, and these are shown by default at the very top or very bottom of the view.
You can use click and drag to either navigate around the region or highlight regions of interest, Click on the Drag/Select option at the top or bottom right to switch mouse action. On Drag, you can click and drag left or right to move along the genome, the page will reload when you drop the mouse button. On Select you can drag out a box to highlight or zoom in on a region of interest.
With the tool set to Select, drag out a box around an exon and choose Mark region.
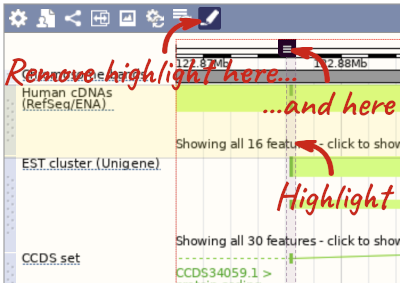
The highlight will remain in place if you zoom in and out or move around the region. This allows you to keep track of regions or features of interest.
We can edit what we see on this page by clicking on the blue Configure this page menu at the left.

This will open a menu that allows you to change the image.
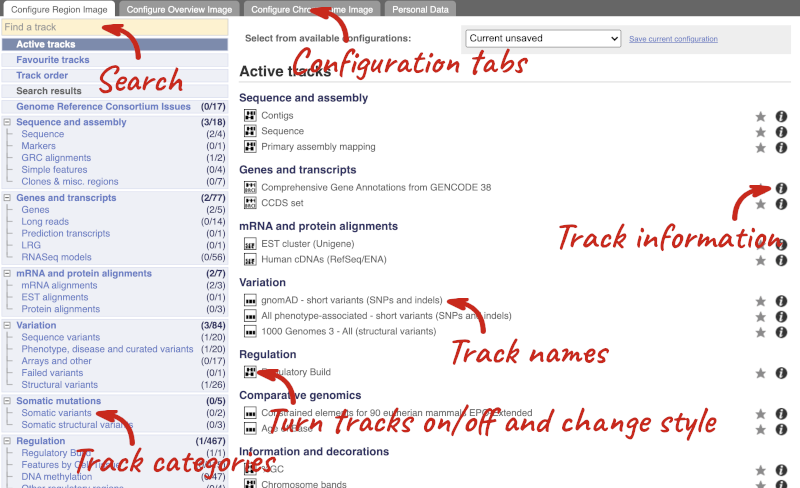
There are thousands of possible tracks that you can add. When you launch the view, you will see all the tracks that are currently turned on with their names on the left and an info icon on the right, which you can click on to expand the description of the track. Turn them on or off, or change the track style by clicking on the box next to the name. More details about the different track styles are in this FAQ: http://www.ensembl.org/Help/Faq?id=335.
You can find more tracks to add by either exploring the categories on the left, or using the Find a track option at the top left. Type in a word or phrase to find tracks with it in the track name or description.
Let’s add some tracks to this image. Add:
- Proteins (mammal) from UniProt – Labels
- 1000 Genomes - All - short variants (SNPs and indels) – Normal
Now click on the tick in the top left hand to save and close the menu. Alternatively, click anywhere outside of the menu. We can now see the tracks in the image. The proteins track is stranded, so you will see two tracks, one above and one below the contig, representing the proteins mapped to the forward and reverse strands respectively. The variants track is not stranded, so is found near the bottom of the image.
If the track is not giving you can information you need, you can easily change the way the tracks appear by hovering over the track name then the cog wheel to open a menu. To make it easier to compare information between tracks, such as spotting overlaps, you can move tracks around by clicking and dragging on the bar to the left of the track name.

Now that you’ve got the view how you want it, you might like to show something you’ve found to a colleague or collaborator. Click on the Share this page button to generate a link. Email the link to someone else, so that they can see the same view as you, including all the tracks you’ve added. These links contain the Ensembl release number, so if a new release or even assembly comes out, your link will just take you to the archive site for the release it was made on.
To return this to the default view, go to Configure this page and select Reset configuration at the bottom of the menu.
Genes and transcripts
You can find out lots of information about Ensembl genes and transcripts using the browser. If you’re already looking at a region view, you can click on any transcript and a pop-up menu will appear, allowing you to jump directly to that gene or transcript.
Alternatively, you can find a gene by searching for it. You can search for gene names or identifiers, and also phenotypes or functions that might be associated with the genes.
We’re going to look at the human UQCRQ gene. From ensembl.org, type UQCRQ into the search bar and click the Go button. You will get a list of hits with the human gene at the top.
Where you search for something without specifying the species, or where the ID is not restricted to a single species, the most popular species will appear first, in this case, human, mouse and zebrafish appear first. You can restrict your query to species or features of interest using the options on the left.
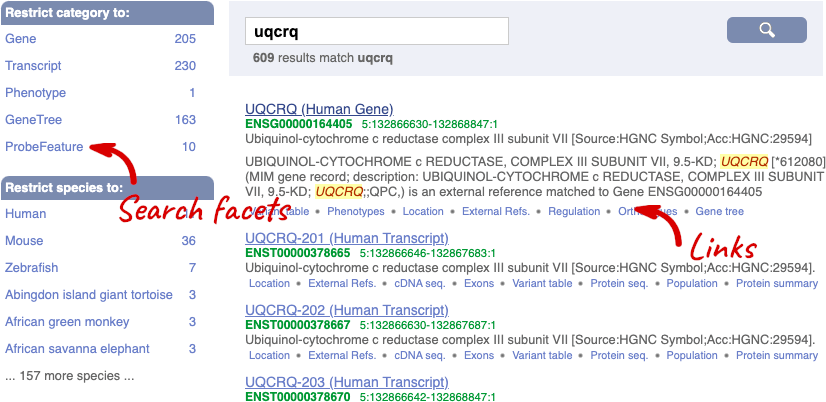
The gene tab
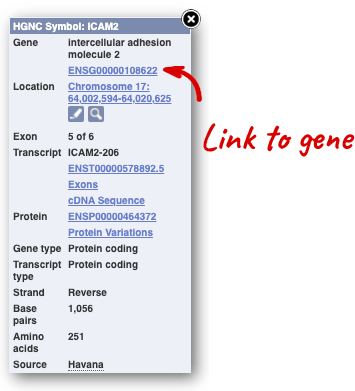
Click on the gene name or Ensembl ID. The Gene tab should open:
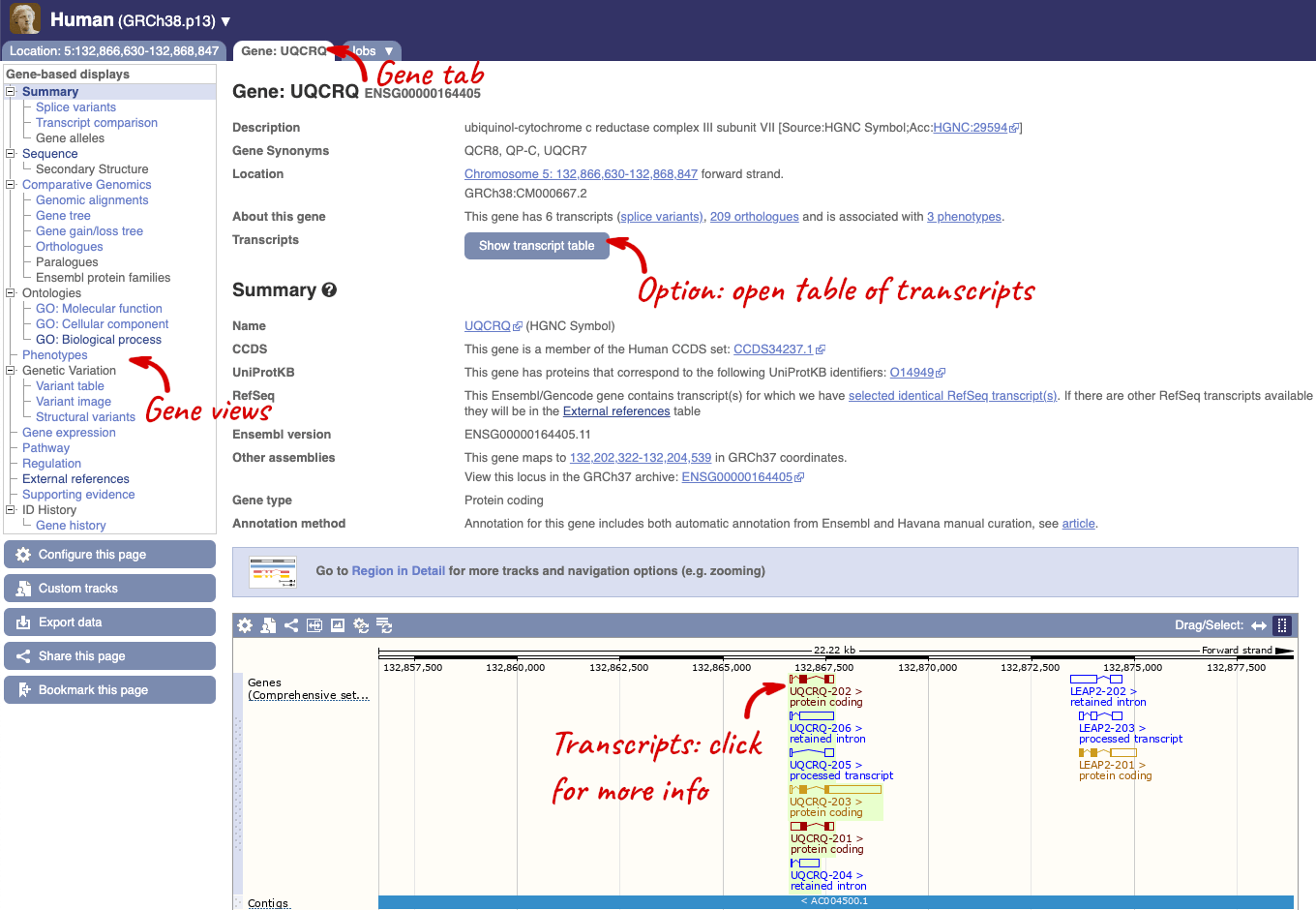
This page summarises the gene, including its location, name and equivalents in other databases. At the bottom of the page, a graphic shows a region view with the transcripts. We can see exons shown as blocks with introns as lines linking them together. Coding exons are filled, whereas non-coding exons are empty. We can also see the overlapping and neighbouring genes and other genomic features.
There are different tabs for different types of features, such as genes, transcripts or variants. These appear side-by-side across the blue bar, allowing you to jump back and forth between features of interest. Each tab has its own navigation column down the left hand side of the page, listing all the things you can see for this feature.
Let’s walk through this menu for the gene tab. How can we view the genomic sequence? Click Sequence at the left of the page.
The sequence is shown in FASTA format. The FASTA header contains the genome assembly, chromosome, coordinates and strand (1 or -1) – this gene is on the positive strand.
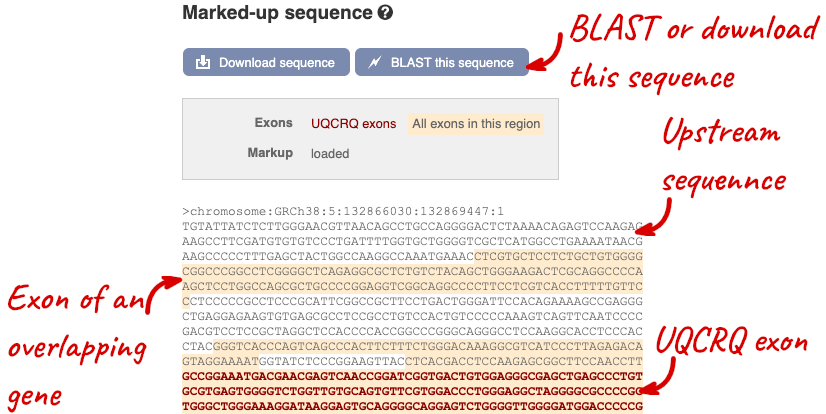
Exons are highlighted within the genomic sequence, both exons of our gene of interest and any neighbouring or overlapping gene. By default, 600 bases are shown up and downstream of the gene. We can make changes to how this sequence appears with the blue Configure this page button found at the left. This allows us to change the flanking regions, add variants, add line numbering and more. Click on it now.
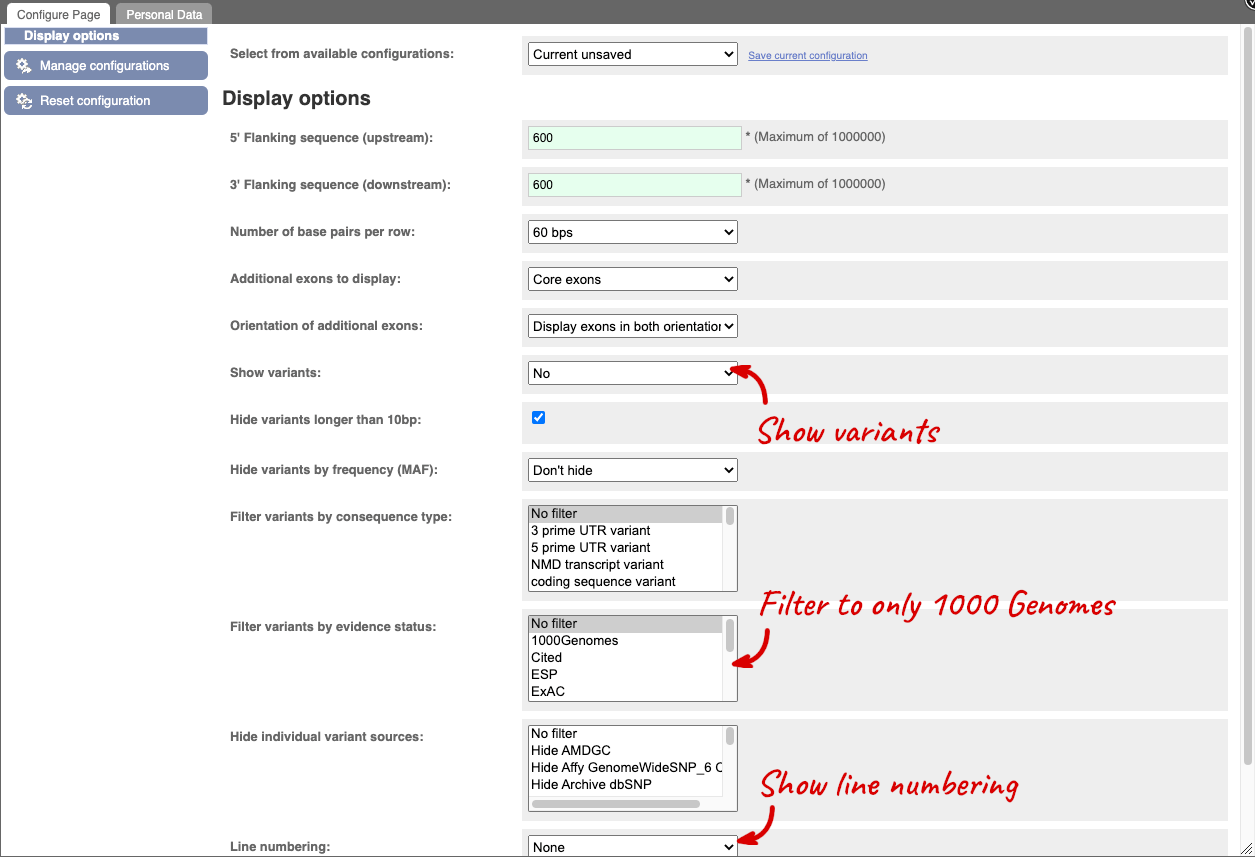
Once you have selected changes (in this example, Show variants, 1000 Genomes variants and Line numbering) click at the top right.
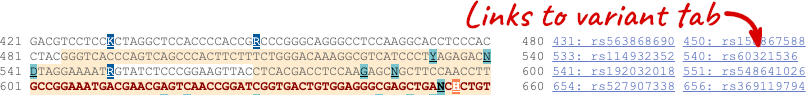
You can download this sequence by clicking in the Download sequence button above the sequence:

This will open a dialogue box that allows you to pick between plain FASTA sequence, or sequence in RTF, which includes all the coloured annotations and can be opened in a word processor. If you want run a sequence analysis tool, download as FASTA sequence, whereas if you want to analyse the sequence visually, RTF is best for this. This button is available for all sequence views.
To find out what the protein does, have a look at GO terms from the Gene Ontology consortium. There are three pages of GO terms, representing the three divisions in GO: Biological process (what the protein does), Cellular component (where the protein is) and Molecular function (how it does it). Click on GO: Biological process to see an example of the GO pages.

Here you can see the functions that have been associated with the gene. There are three-letter codes that indicate how the association was made, as well as links to the specific transcript they are linked to.
You can also see the phenotypes associated with a gene. Click on Phenotype in the left hand menu.
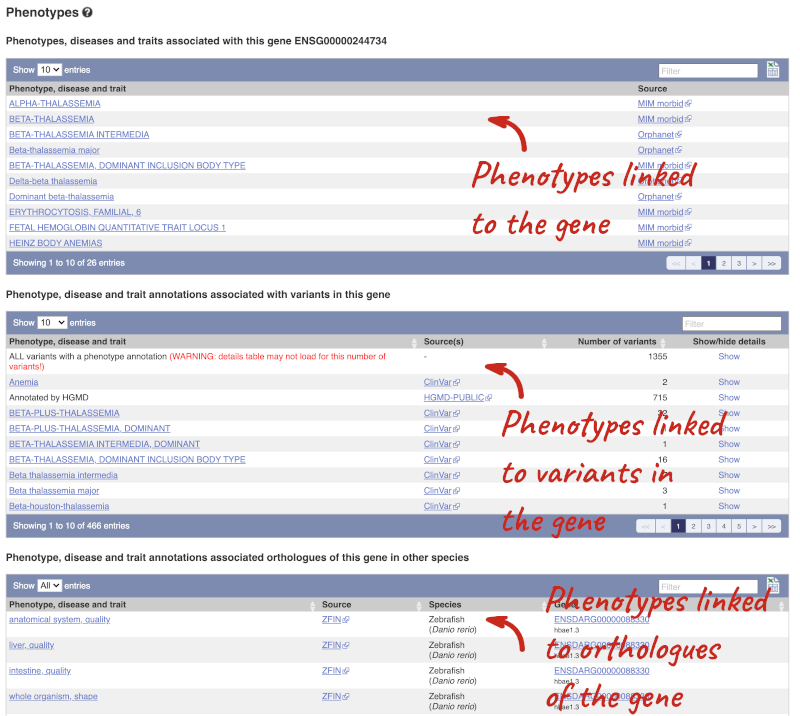
To view all the sequence variants in table form, click the Variant table link at the left of the gene tab.
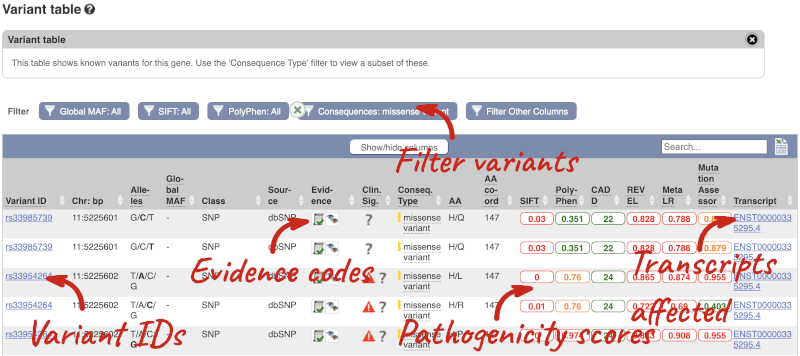
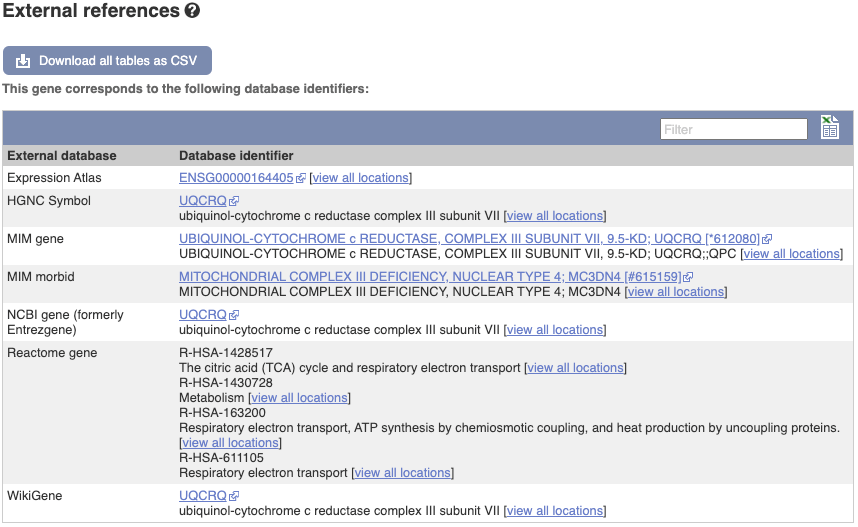
We also have links out to other databases which have information about our genes and may focus on other topics that we don’t cover, like Gene Expression Atlas or OMIM. Go up the left-hand menu to External references:
The transcript tab
We’re now going to explore the different transcripts of UQCRQ. Click on Show transcript table at the top.
![]()
![]()
Here we can see a list of all the transcripts of UQCRQ with their identifiers, lengths, biotypes and flags to help you decide which ones to look at.
If we were to only choose one transcript to analyse, we would choose UQCRQ-203 because it is the MANE Select and Ensembl Canonical. This means it is both 100% identical to the RefSeq transcript NM_014402.5 and both Ensembl and NCBI agree that it is the most biologically important transcript.
Click on the ID, ENST00000378670.8.
You are now in the Transcript tab for UQCRQ-203. We can still see the gene tab so we can easily jump back. The left hand navigation column provides several options for the transcript UQCRQ-203 - many of these are similar to the options you see in the gene tab, but not all of them. If you can’t find the thing you’re looking for, often the solution is to switch tabs.
Click on the Exons link. This page is useful for designing RT-PCR primers because you can see the sequences of the different exons and their lengths.
![]()
You may want to change the display (for example, to show more flanking sequence, or to show full introns). In order to do so click on Configure this page and change the display options accordingly.
Now click on the cDNA link to see the spliced transcript sequence with the amino acid sequence. This page is useful for mapping between the RNA and protein sequences, particularly genetic variants.
![]()
UnTranslated Regions (UTRs) are highlighted in dark yellow, codons are highlighted in light yellow, and exon sequence is shown in black or blue letters to show exon divides. Sequence variants are represented by highlighted nucleotides and clickable IUPAC codes are above the sequence.
Next, follow the General identifiers link at the left. Just like the External References page in the gene tab, this page shows links out to other databases such as RefSeq, UniProtKB, PDBe and others, this time linked to the transcript or protein product, rather than the gene.
![]()
Exploring a genomic region in human
Go to Ensembl.
-
Go to the region from 32,264,000 to 32,492,000 bp on human chromosome 13. On which cytogenetic band is this region located? How many contigs make up this portion of the assembly (contigs are contiguous stretches of DNA sequence that have been assembled solely based on direct sequencing information)?
-
Zoom in on the BRCA2 gene.
-
Configure this page to turn on the LTR (repeat) track in this view. What tool was used to annotate the LTRs according to the track information? How many LTRs can you see within the BRCA2 gene? Do any overlap exons?
-
Create a Share link for this display. Email it to your neighbour. Open the link they sent you and compare. If there are differences, can you work out why?
-
Export the genomic sequence of the region you are looking at in FASTA format.
-
Turn off all tracks you added to the Region in detail page.
- Go to the Ensembl homepage, select Human from the Species drop-down list and type
13:32264000-32492000in the text box (alternatively leave the Search drop-down list as it is and type13:32264000-324920000in the text box). Click Go.This genomic region is located on cytogenetic band q13.1. It is made up of three contigs, indicated by the alternating light and dark blue coloured bars in the Contigs track.
-
Draw with your mouse a box encompassing the BRCA2 transcripts. Click on Jump to region in the pop-up menu.
- Click Configure this page in the side menu (or on the cog wheel icon in the top left hand side of the bottom image). Go into Repeats in the left-hand menu then select LTR. Click on the (i) button to find out more information.
Repeat Masker was used to annotate LTRs onto the genome.
Save and close the new configuration by clicking on ✓ (or anywhere outside the pop-up window). There are ten LTRs overlapping BRCA2, none of them overlap exons. -
Click Share this page in the side menu. Copy the URL. Get your neighbour’s email address and compose an email to them, paste the link in and send the message. When you receive the link from them, open the email and click on your link. You should be able to view the page with the new configuration and data tracks they have added to in the Location tab. You might see differences where they specified a slightly different region to you, or where they have added different tracks.
Here is the Share link from the video answer: https://may2021.archive.ensembl.org/Homo_sapiens/Share/71a173bba78f0dbe03e48d3240424943?redirect=no;mobileredirect=no
-
Click Export data in the side menu. Leave the default parameters as they are (FASTA sequence should already be selected). Click Next>. Click on Text. Note that the sequence has a header that provides information about the genome assembly (GRCh38), the chromosome, the start and end coordinates and the strand. For example:
>13_dna:chromosome_chromosome:GRCh38:13:32311910:32405865:1 - Click Configure this page in the side menu. Click Reset configuration. Click ✓.
Exploring the MYH9 gene in human
- In Ensembl, find the human MYH9 (myosin, heavy chain 9, non-muscle) gene and open the Gene tab.
- On which chromosome and which strand of the genome is this gene located?
- How many transcripts (splice variants) are there and how many are protein coding?
- What is the longest protein-coding transcript, and how long is the protein it encodes?
- Which transcript would you take forward for further study?
-
Click on Phenotypes at the left side of the page. Are there any diseases associated with this gene, according to Mendelian Inheritance in Man (MIM)?
-
What are some functions of MYH9 according to the Gene Ontology (GO) consortium? Have a look at the GO: Biological process pages for this gene.
- In the transcript table, click on the transcript ID for MYH9-201, and go to the Transcript tab.
- How many exons does it have?
- Are any of the exons completely or partially untranslated?
- Is there an associated sequence in UniProtKB/Swiss-Prot? Have a look at the General identifiers for this transcript.
- Are there microarray (oligo) probes that can be used to monitor ENST00000216181 expression?
- Select Human from the Species drop-down list and type
MYH9. Click Go. Click on MYH9 (Human Gene) in the search results which will send you to the Gene tab.- The gene is located on chromosome 22 on the reverse strand.
- Ensembl has 23 transcripts annotated for this gene, of which 6 are protein-coding.
- The longest protein-coding transcript is MYH9-215 and it codes for a protein that is 1,981 amino acids long.
- MYH9-201 is the transcript I would take forward for further study, as it is the MANE Select transcript (for a description, mouse-over the MANE Select flag in the transcript table).
- Click on Phenotypes in the left-hand panel to see the associated phenotypes. There is a large table of phenotypes. To see only the ones from MIM, type
MIMinto the filter box at the top right-hand corner of the table.These are some of the phenotypes associated with MYH9 according to MIM: Deafness, Autosomal dominant 17 and Macrothrombocytopenia and granulocyte inclusions with or without nephritis or sensorineural hearing loss. You can click on the records for more information.
-
The Gene Ontology project maps terms to a protein in three classes: biological process, cellular component, and molecular function. Click on GO: Biological process on the left-hand panel. Angiogenesis, cell adhesion, and protein transport are some of the roles associated with MYH9. All GO terms are associated with a single transcript: ENST00000216181.
- Click on ENST00000216181.11 in the transcript table. You should now be on the Transcript tab.
- It has 41 exons, shown in the Transcript summary.
Click on the Exons link in the left-hand panel.
- Exon 1 is completely untranslated, and exons 2 and 41 are partially untranslated (UTR sequence is shown in orange). You can also see this in the cDNA view if you click on the cDNA link in the left side menu.
Click on General identifiers in the left-hand panel.
- P35579.254 from UniProt/Swiss-Prot matches the translation of the Ensembl transcript. Click on P35579.254 to go to UniProtKB, or click align for the alignment.
- Click on Oligo probes in the left-hand panel.
Probesets from Affymetrix, Agilent, Codelink, Illumina, and Phalanx OneArray match to this transcript sequence. Expression analysis with any of these probesets would reveal information about the transcript. Hint: this information can sometimes be found in the [ArrayExpress Atlas] (https://www.ebi.ac.uk/biostudies/arrayexpress).
Finding a gene associated with a phenotype
Phenylketonuria is a genetic disorder caused by an inability to metabolise phenylalanine in any body tissue. This results in an accumulation of phenylalanine causing seizures and intellectual disability.
(a) Search for phenylketonuria from the Ensembl homepage and narrow down your search to only genes. What gene is associated with this disorder?
(b) How many protein coding transcripts does this gene have? View all of these in the transcript comparison view.
(c) What is the MIM gene identifier for this gene?
(d) Go to the MANE Select transcript and look at its 3D structure. In the model 2pah, how many protein molecules can you see?
(a) Start at the Ensembl homepage (http://www.ensembl.org).
Type phenylketonuria into the search box then click Go. Choose Gene from the left hand menu.
The gene associated with this disorder is PAH, phenylalanine hydroxylase, ENSG00000171759.
(b) If the transcript table is hidden, click on Show transcript table to see it.
There are six protein coding transcripts.
Click on Transcript comparison in the left hand menu. Click on Select transcripts. Either select all the transcripts labelled protein coding one-by-one, or click on the drop down and select Protein coding. Close the menu.
(c) Click on External references.
The MIM gene ID is 612349.
(d) Open the transcript table and click on the ID for the MANE Select: ENST00000553106.6. Go to PDB 3D protein model in the left-hand menu.
The model 2pah is shown by default. It has two protein molecules in it. You may need to rotate the model to see this clearly.