Filter Events by Year
Ensembl Browser Workshop: Genomics for Dermatology
Course Details
- Lead Trainer
- Louisse Paola Mirabueno
- Event Date
- 2022-11-29
- Location
- Hinxton Hall Conference Centre, Wellcome Genome Campus, Hinxton, UK
- Description
- Work with the Ensembl Outreach team to get hands-on experience accessing and analysing variation data with the Ensembl genome browser.
Demos and exercises
Variation
In any of the sequence views shown in the Gene and Transcript tabs, you can view variants on the sequence. You can do this by clicking on Configure this page from any of these views.
Let’s take a look at the Gene sequence view for HBB in human. Search for HBB and go to the Sequence view.
If you can’t see variants marked on this view, click on Configure this page and select Show variants: Yes and show links. You may also wish to add a filter to the variants to allow them to load more quickly, we’ll add Filter variants by evidence status: 1000Genomes.
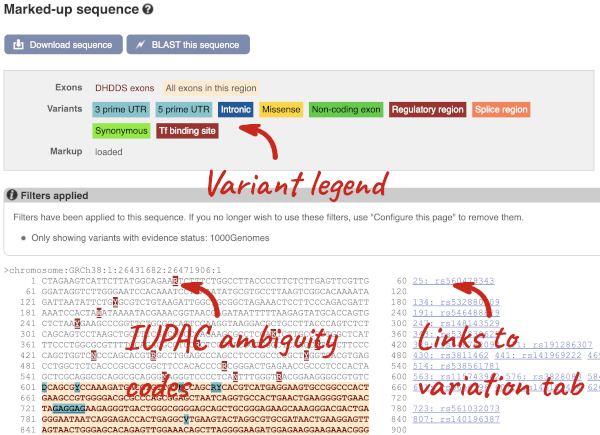
Find out more about a variant by clicking on it.
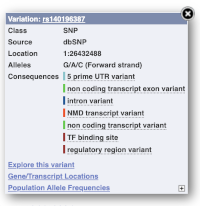
You can add variants to all other sequence views in the same way.
You can go to the Variation tab by clicking on the variant ID. For now, we’ll explore more ways of finding variants.
To view all the sequence variants in table form, click the Variant table link at the left of the gene tab.
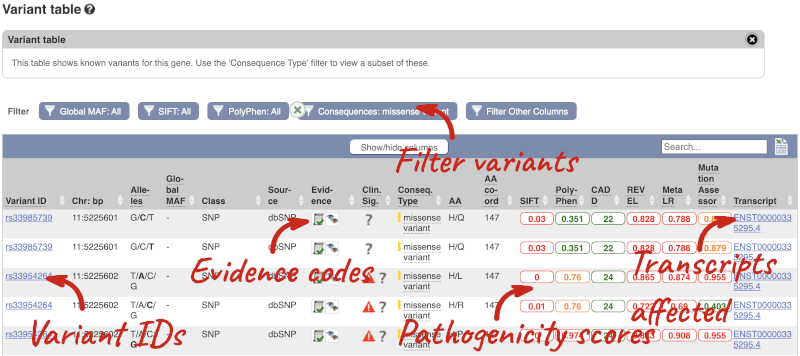
You can filter the table to only show the variants you’re interested in. For example, click on Consequences: All, then select the variant consequences you’re interested in. For display purposes, the table above has already been filtered to only show missense variants.
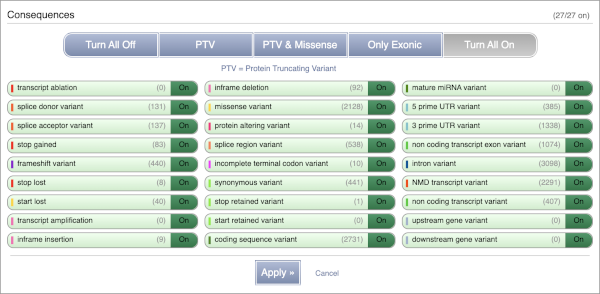
You can also filter by the different pathogenicity scores and MAF, or click on Filter other columns for filtering by other columns such as Evidence or Class.
The table contains lots of information about the variants. You can click on the IDs here to go to the Variation tab too.
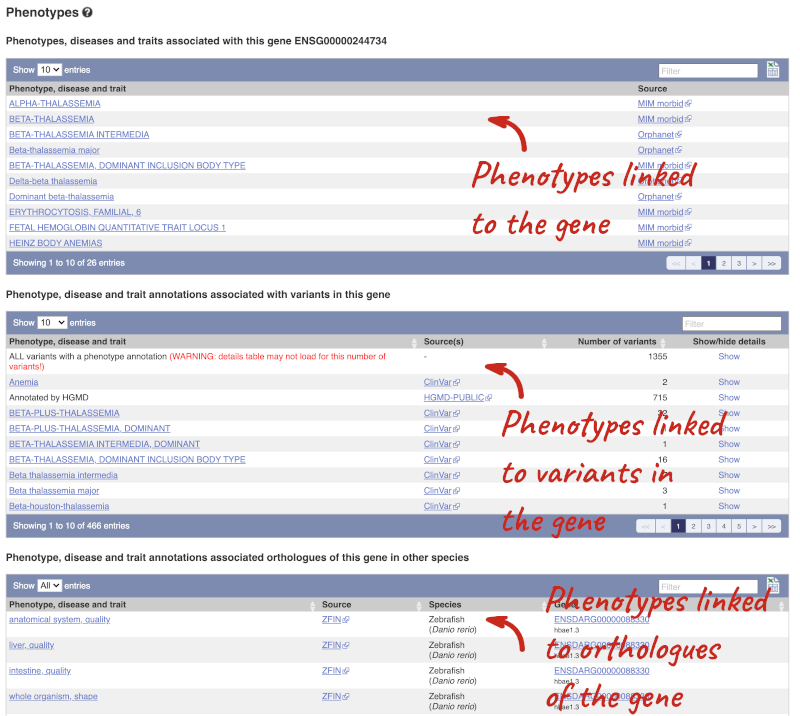
You can also see the phenotypes associated with a gene. Click on Phenotype in the left hand menu.
Open the transcript table and go to HBB-201 ENST00000335295, then click on Haplotypes in the left hand menu.
![]()
The Haplotypes view in the transcript tab shows you the actual protein and CDS sequences in 1000 Genomes individuals. This is possible because the 1000 Genomes study has phased genotypes, so we know which alleles occur on which of the chromosome pairs. The table lists all the versions of the protein that occur along with their frequencies, including the reference sequence and sequences with one or more alternative alleles.
Click on one of the haplotypes, we’ll go for 18K>*,19del{130}, to find out more about it. Here you will see the frequency in the 1000 Genomes subpopulations, the sequence and the 1000 Genomes individuals where this protein is found.
Let’s have a look at variants in the Location tab. Click on the Location tab in the top bar.
Configure this page and open Variation from the left-hand menu.
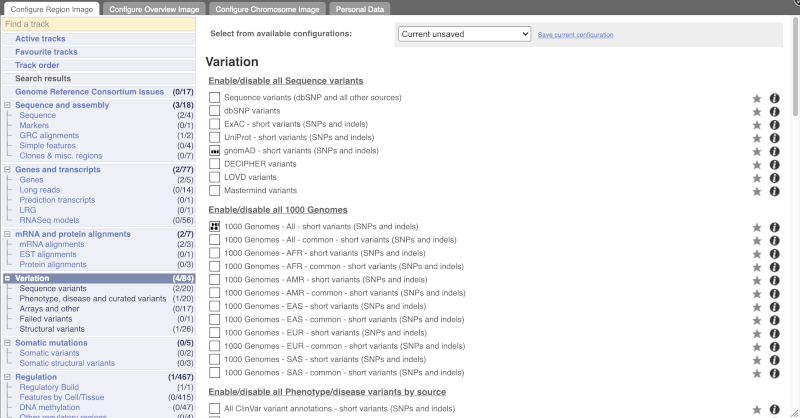
There are various options for turning on variants. You can turn on variants by source, by frequency, presence of a phenotype or by individual genome they were isolated from. You can also turn on genotyping chips.
Let’s have a look at a specific variant. If we zoomed in we could see the variant rs334 in this region, however it’s easier to find if we put rs334 into the search box. Click through to open the Variation tab.
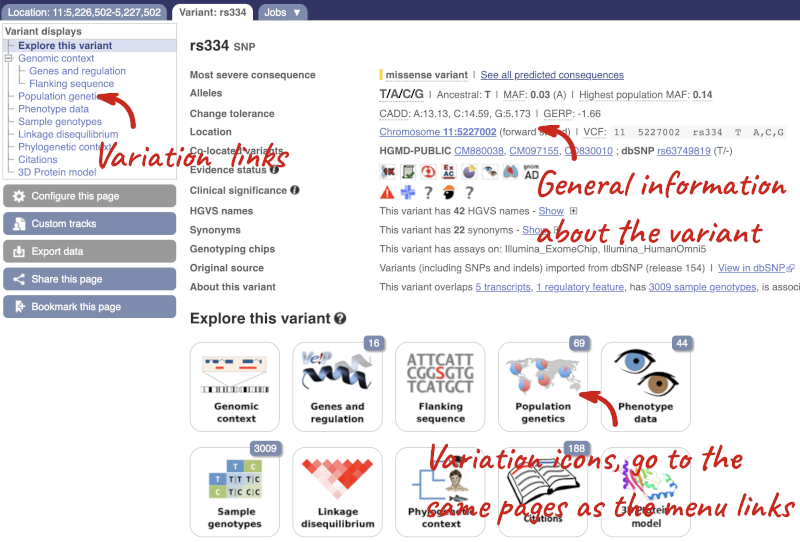
The icons show you what information is available for this variant. Click on Genes and regulation, or follow the link on the left.
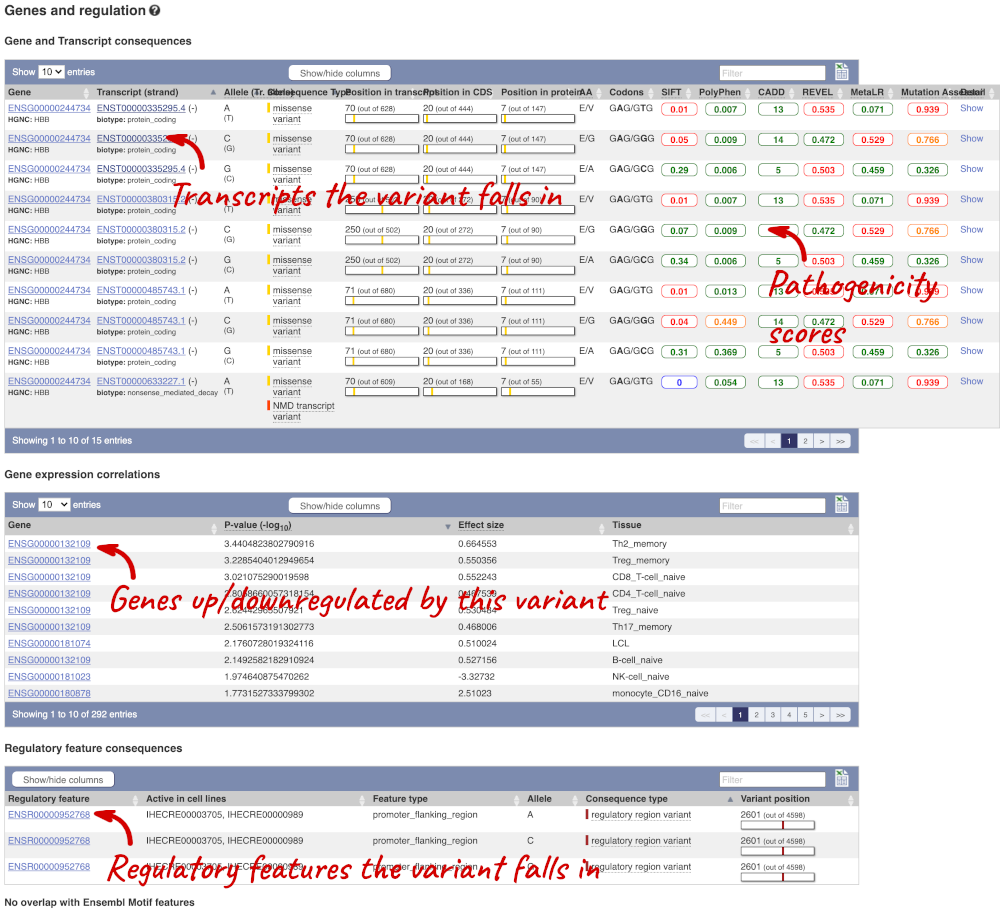
This page illustrates the genes the variant falls within and the consequences on those genes, including pathogenicity predictors. It also shows data from GTEx on genes that have increased/decreased expression in individuals with this variant, in different tissues. Finally, regulatory features and motifs that the variant falls within are shown.
We can also see the variant in the protein structure by clicking on 3D Protein model.
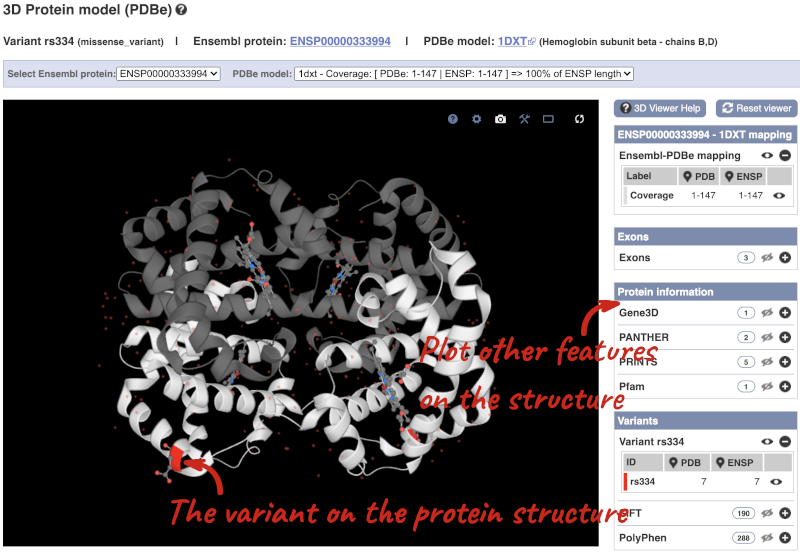
This is a LiteMol viewer, where you can rotate and zoom in on the structure. The variant location is highlighted, so you can see where it lands within the structure.
Let’s look at population genetics. Click on Population genetics in the left-hand menu.
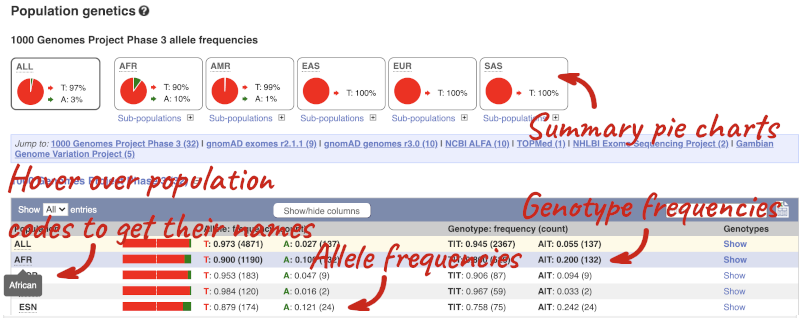
The population allele frequencies are shown by study, including 1000 Genomes and gnomAD. Where genotype frequencies are available, these are shown in the tables.
There are big differences in allele frequencies between populations. Let’s have a look at the phenotypes associated with this variant to see if they are known to be specific to certain human populations. Click on Phenotype Data in the left-hand menu.
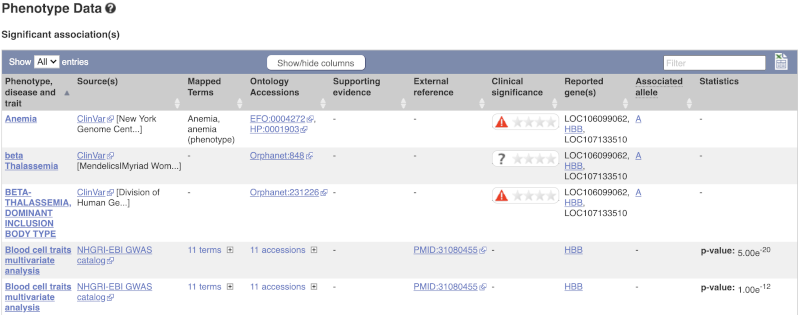
This variant is associated with various phenotypes, including sickle cell and malaria resistance. These phenotype associations come from sources including the GWAS catalog, ClinVar, Orphanet and OMIM. Where available, there are links to the original paper that made the association, the allele that is associated with the phenotype and p-values and other statistics.
Exploring a SNP in human
The missense variation rs61746355 in the human KRT6B gene has been reported in patients with delayed‐onset pachyonychia congenita (PC). This SNP is also referred to as ‘c.1495G>A’ or ‘p.Gly499Ser’.
(a) Find the page with information for rs61746355.
(b) What sequence ontology (SO) terms, or variant consequence types, are associated with rs61746355? What are the amino acid changes?
(c) Why are the alleles for this variant in Ensembl given as C/A and not as G/T, as in the literature?
(d) What is the major allele of rs61746355 in different populations?
(e) In which paper(s) is the association between rs61746355 and PC described?
(f) According to the data imported from dbSNP, the ancestral allele for rs61746355 is C. Ancestral alleles in dbSNP are based on a comparison between human and chimp. Does the sequence at this same position in other primates confirm that the ancestral allele is C?
(a) Go to the Ensembl homepage.
Type rs61746355 in the Search box, then click Go.
(b) Click on Genes and Regulation in the left-hand menu (or the Genes and Regulation icon under Explore this variant).
The rs61746355 variant is a missense variant. Please note that this variant is multi-alleleic with two alternative alleles A and C. The table displays one consequence per row, with each transcript being listed twice. The amino acid change is G/C for allele A, and G/S for allele T.
(c) In Ensembl, the alleles of rs61746355 are given as C/A/T. You can find this at the top of the page under Alleles.
This is because these are the alleles in the forward strand of the genome. In the literature, the alleles are given as G/T/A because the KRT6B gene is located on the reverse strand. The alleles in the actual gene and transcript sequences are G/T/A. In Ensembl, the allele that is present in the reference genome assembly is always put first.
(d) Click on Population genetics in the side menu.
In all populations, the allele C is the major one.
(e) Click on Phenotype Data in the left hand side menu.
The specific studies where the association was originally described is given in Phenotype data in the left-hand menu. A link between rs61746355 and PC was described in 1 paper. Click on the PubMed ID PMID:24354895 to view the publication.
(f) Click on Phylogenetic Context in the left-hand menu. Select Alignment: 10 primates EPO and click Apply.
Bonobo, chimpanzee, crab-eating macaque, gibbon, gorilla, macaque, mouse lemur and sumatran orangutan all have a C allele in this position. Chlorocebus sabaeus (vervet-AGM) has an A allele at this position.
Exploring sequence variant annotation in human
The NF1 gene encodes a neurofibromin 1 protein. Variations in the gene have been associated with neurofibromatosis, a disease characterized by patches of skin pigmentation (also known as café-au-lait spots).
(a) Find the NF1 gene for human. How many variants are listed for this gene? How many structural variants are listed for this gene?
(b) Let’s focus on the Matched Annotation between NCBI and EBI (MANE) Select transcript of the NF1 gene. The MANE Select transcript flag describes the confidence of the transcript annotation. You can read more about this here. Find the rs876658658 variant. What Sequence Ontology (SO) term, or variant consequence type, has been assigned to this variant?
(c) What is the clinical significance for this variant?
(d) Why does Ensembl put the C allele first (C/T)?
(e) What is the ancestral allele predicted for this locus?
(f) Which allele is associated with type 1 neurofibromatosis (with supporting evidence) and what is the significance of the association?
(g) How many publications mention this variant?
(h) Besides neurofibromatosis, what other MIM morbid entries are available for this gene?
(a) Go to the Ensembl homepage, search for NF1 in human and click on the first hit “NF1 (Human Gene)” in the results page. Click on Variant table in the left-hand menu.
A notice in a yellow box will show you that there are 76,692 variants for this gene.
Now click on _Structural variants_ in the left-hand menu and scroll to the bottom of the table. >There are 966 entries for structural variants.
(b) Under Show transcript table, click on ENST00000358273.9. This opens the Transcript tab. Now go to Variant table on the left-hand menu and search for rs876658658 in the table.
The rs876658658 has a ‘stop gained’ SO term assigned to it.
(c) In the Variant table, look for the Clin. Sig. column.
The rs876658658 variants is described to be as both ‘likely pathogenic’ and ‘pathogenic’.
(d) In Ensembl, the allele that is present in the reference genome assembly is put first, i.e. C.
Usually in literature, the major allele (in the population of interest) is put first. In the case of rs876658658 the allele in the reference genome is the major allele, but as the reference genome is a mosaic of the genomes of just a few individuals this is by no means the case for all variants.
(e) Click on the variant rs876658658. This opens the Variant tab. Focus on the summary information at the top of the page.
The ancestral allele is reported as C, which you can find in the Alleles line.
(f) Click on Phenotype Data in the left-hand menu.
The T allele was reported to be associated with type 1 neurofibromatosis with supporting evidence.
(g) Click on Citations in the left-hand menu.
3 publications mention this variant.
(h) Switch to the Gene: NF1 at the top of the page. Click on Phenotypes in the left-hand menu. You can filter the Phenotypes, diseases and traits associated with this gene ENSG00000196712 table by entering “MIM morbid” in the text field at the top right corner of the table.
Other MIM morbid entries for the NF1 include Neurofibromatosis-Noonan syndrome and Watson syndrome.
VEP
We have identified five variants on human chromosome nine, C-> A at 128203516, an A deletion at 128328461, C->A at 128322349, C->G at 128323079 and G->A at 128322917.
We will use the Ensembl VEP to determine:
- Have my variants already been annotated in Ensembl?
- What genes are affected by my variants?
- Do any of my variants affect gene regulation?
Go to the front page of Ensembl and click on the Variant Effect Predictor.

This page contains information about the VEP, including links to download the script version of the tool. Click on Launch VEP to open the input form:
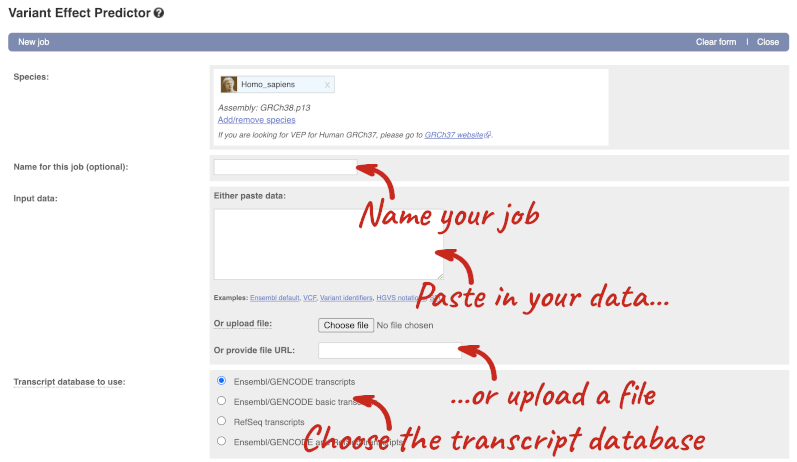
The data is in VCF format:
chromosome coordinate id reference alternative
Put the following into the Paste data box:
9 128328460 var1 TA T
9 128322349 var2 C A
9 128323079 var3 C G
9 128322917 var4 G A
9 128203516 var5 C A
The VEP will automatically detect that the data is in VCF.
There are further options that you can choose for your output. These are categorised as Identifiers, Variants and frequency data, Additional annotations, Predictions, Filtering options and Advanced options. Let’s open all the menus and take a look.
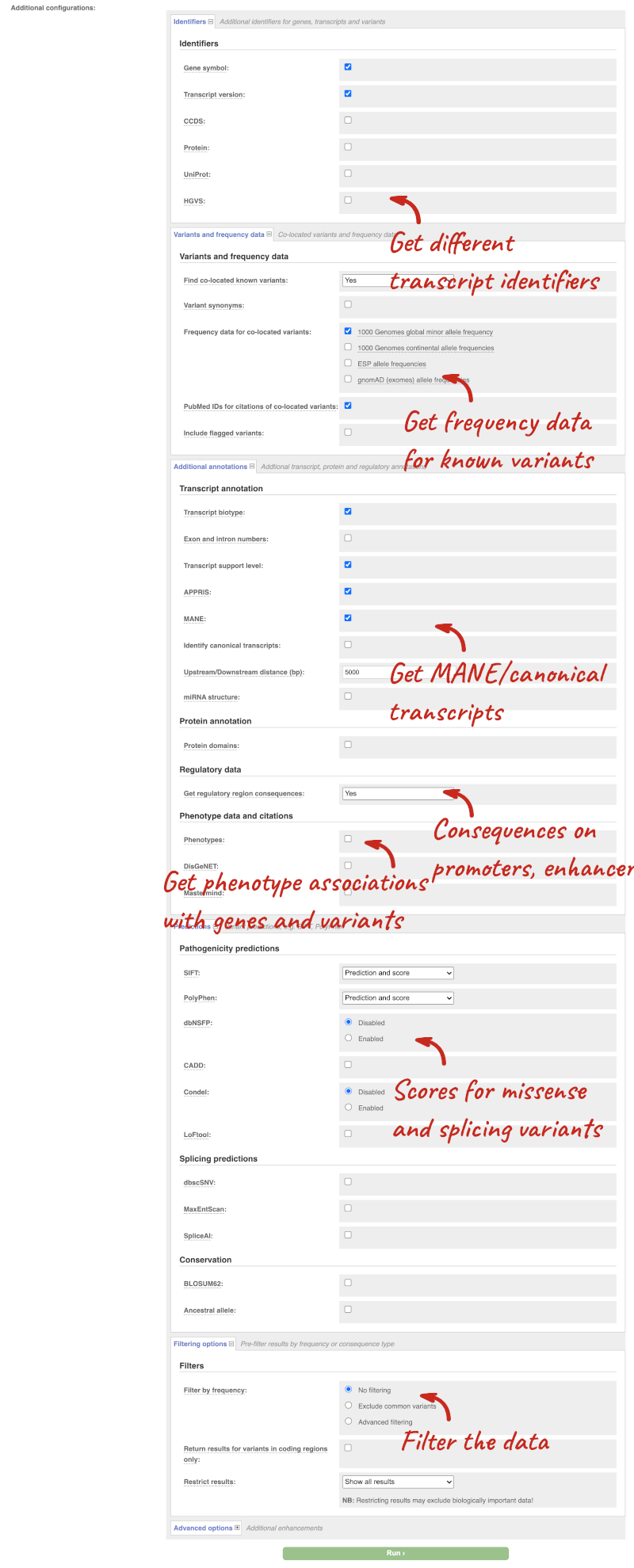
Hover over the options to see definitions.
We’re going to select some options:
- HGVS, annotation of variants in terms of the transcripts and proteins they affect, commonly-used by the clinical community
- Phenotypes
- Protein domains
When you’ve selected everything you need, scroll right to the bottom and click Run.

The display will show you the status of your job. It will say Queued, then automatically switch to Done when the job is done, you do not need to refresh the page. You can edit or discard your job at this time. If you have submitted multiple jobs, they will all appear here.
Click View results once your job is done.
In your results you will see a graphical summary of your data, as well as a table of your results.
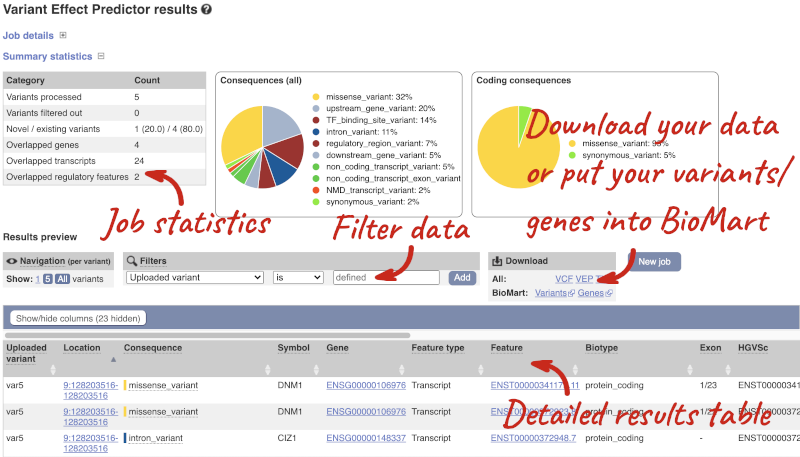
The results table is enormous and detailed, so we’re going to go through the it by section. The first column is Uploaded variant. If your input data contains IDs, like ours does, the ID is listed here. If your input data is only loci, this column will contain the locus and alleles of the variant. You’ll notice that the variants are not neccessarily in the order they were in in your input. You’ll also see that there are multiple lines in the table for each variant, with each line representing one transcript or other feature the variant affects.
You can mouse over any column name to get a definition of what is shown.
The next few columns give the information about the feature the variant affects, including the consequence. Where the feature is a transcript, you will see the gene symbol and stable ID and the transcript stable ID and biotype. Where the feature is a regulatory feature, you will get the stable ID and type. For a transcription factor binding motif (labelled as a MotifFeature) you will see just the ID. Most of the IDs are links to take you to the gene, transcript or regulatory feature homepage.
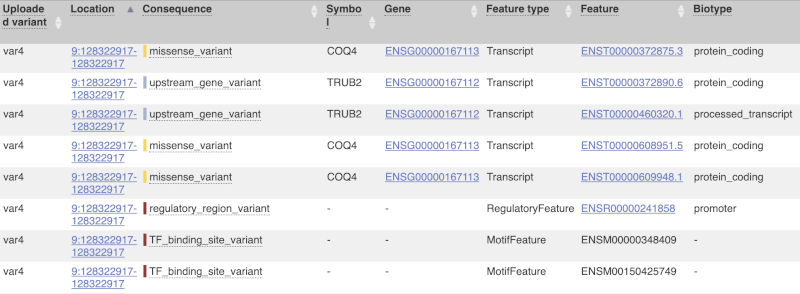
This is followed by details on the effects on transcripts, including the position of the variant in terms of the exon number, cDNA, CDS and protein, the amino acid and codon change, transcript flags, such as MANE, on the transcript, which can be used to choose a single transcript for variant reporting, and predicted pathogenicity scores. The predicted pathogenicity scores are shown as numbers with coloured highlights to indicate the prediction, and you can mouse-over the scores to get the prediction in words. Two options that we selected in the input form are the HGVS notation, which is shown to the left of the image below and can be used for reporting, and the Domains to the right. The Domains list the proteins domains found, and where there is available, provide a link to the 3D protein model which will launch a LiteMol viewer, highlighting the variant position.
![]()
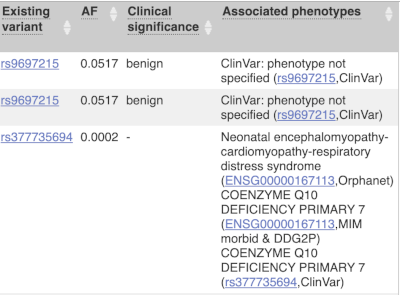
Where the variant is known, the ID of the existing variant is listed, with a link out to the variant homepage. In this example, only rsIDs from dbSNP are shown, but sometimes you will see IDs from other sources such as COSMIC. The VEP also looks up the variant from frequency files, and pulls back the allele frequency (AF in the table), which will give you the 1000 Genomes Global Allele Frequency. In our query, we have not selected allele frequencies from the continental 1000 Genomes populations or from gnomAD, but these could also be shown here. We can also see ClinVar clinical significance and the phenotypes associated with known variants or with the genes affected by the variants, with the variant ID listed for variant associations and the gene ID listed for gene associations, along with the source of the association.
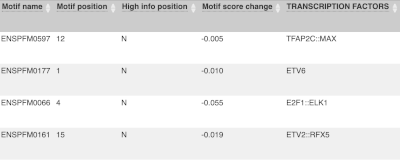
For variants that affect transcription factor binding motifs, there are columns that show the effect on motifs (you may need to click on Show/hide columns at the top left of the table to display these). Here you can see the position of the variant in the motif, if the change increases or decreases the binding affinity of the motif and the transcription factors that bind the motif.
Above the table is the Filter option, which allows you to filter by any column in the table. You can select a column from the drop-down, then a logic option from the next drop-down, then type in your filter to the following box. We’ll try a filter of Consequence, followed by is then missense_variant, which will give us only variants that change the amino acid sequence of the protein. You’ll notice that as you type missense_variant, the VEP will make suggestions for an autocomplete.
You can export your VEP results in various formats, including VCF. When you export as VCF, you’ll get all the VEP annotation listed under CSQ in the INFO column. After filtering your data, you’ll see that you have the option to export only the filtered data. You can also drop all the genes you’ve found into the Gene BioMart, or all the known variants into the Variation BioMart to export further information about them.
VEP analysis of FLG variants in human
Below, you will find a list of variants which have been reported in the FLG gene (ENSG00000143631), which codes for filaggrin. Mutations in the gene have been associated with atopic dermatitis.
- C/A at 1: 152,302,797
- G/A at 1: 152,307,085
- C/T at 1: 152,310,208
- G/A at 1: 152,308,234
- C/T at 1: 152,313,454
- C/T at 1: 152,309,920
- A/G at 1: 152,309,268
Use the VEP tool in Ensembl and find out the following information:
-
What consequences have been predicted for the variants?
-
Are SIFT and PolyPhen predictions available for the variants? What are the scores?
-
How many of these variants have been previously reported?
Go to the Ensembl homepage and click on the link VEP at the top of the page. Launch the VEP web interface. You will need to convert your variants into one of VEP’s supported input formats. We have converted the variants into the Ensembl default format below. Paste the variants into Input data:.
1 152302797 152302797 C/A
1 152307085 152307085 G/A
1 152310208 152310208 C/T
1 152308234 152308234 G/A
1 152313454 152313454 C/T
1 152309920 152309920 C/T
1 152309268 152309268 A/G
Enable SIFT and PolyPhen score predictions in the Predictions under Additional configurations and click Run. Once your job is done, click View results. You will retrieve a table of your VEP results. Note that there may be multiple entries in the table for each variant. VEP will give you all consequences for each feature the variant falls within.
- Filter your table to show variants affecting the FLG gene (ENSG00000143631) only. Under Filters select Gene from the drop-down menu and enter
ENSG00000143631in the text field. Click Add.Look for the Consequence column. This column will give you the sequence ontology (SO) terms (i.e. synonymous, missense, downstream, intronic, 5’ UTR, 3’ UTR, etc.) provided by VEP for the listed SNPs. You can read more about the Sequence Ontology project here. All 7 entries have been predicted to have missense consequences.
- Look for the SIFT and PolyPhen columns.
Pathogenicty scores are available for all variants. Benign/tolerated scores are predicted for variants 1_152313454_C/T, 1_152310208_C/T and 1_152309920_C/T. For the other 4 variants, the SIFT and PolyPhen scores do not correspond.
- Look for the Existing variant column. If a variant has been previously described, a link will be available in this column.
4 variants have been previously described: 1_152313454_C/T, 1_152308234_G/A, 1_152310208_C/T and 1_152309920_C/T. Variants 1_152302797_C/A, 1_152309268_A/G and 1_152307085_G/A are novel variants.