Filter Events by Year
Ensembl browser workshop, University of North Carolina at Chapel Hill
Course Details
- Lead Trainer
- Aleena Mushtaq
- Event Dates
- 2021-11-16 until 2021-11-17
- Location
- Virtual
- Description
- Work with the Ensembl Outreach team to get to grips with the Ensembl browser, accessing and analysing genomic data.
- Survey
- Ensembl browser workshop, University of North Carolina at Chapel Hill Feedback Survey
Demos and exercises
Species and genome assemblies
Demo: Introduction to Ensembl
Ensembl
Homepage
The front page of Ensembl is found at ensembl.org. It contains lots of information and links to help you navigate Ensembl:
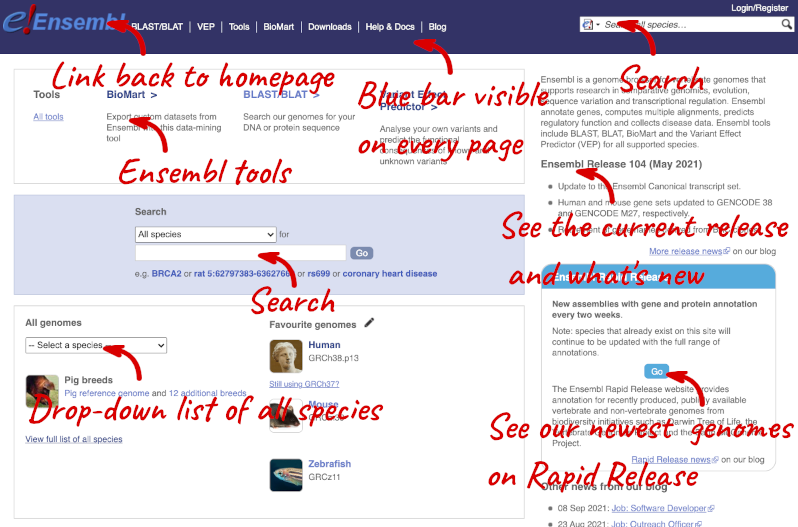
On the right-hand panel you can see the current release number and what has come out in this release. To access old releases, scroll to the bottom of the page and click on View in archive site in the right-hand corner.
Click on the links to go to the archives. Alternatively, you can jump quickly to the correct release by adding e plus the release number in the URL. For example e98.ensembl.org jumps to Ensembl release 98.
Available species
Scroll back up to the top of the homepage. You can view all available species by clicking the View full list of all species link underneath the coloured search block.
You can search for your species of interest (either the common or scientific name) using the search bar at the top right-hand corner of the table. Click on the common name of your species of interest to go to the species information page. We’ll click on Human.
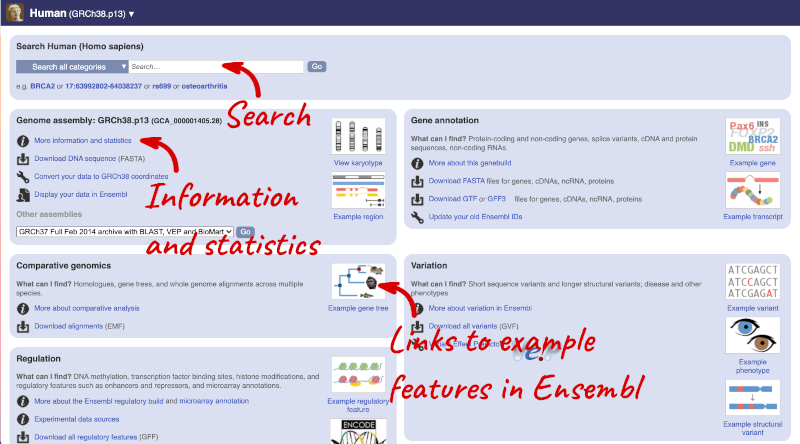
Species information
Here you can see links to example features and to download flatfiles. To find out more about the genome assembly and genebuild, click on More information and statistics under the Genome assembly section.
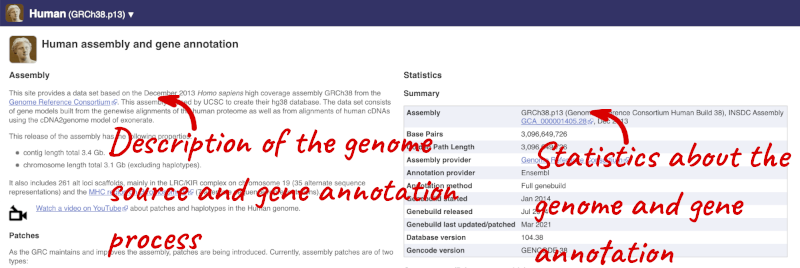
Here you’ll find a detailed description of how to the genome was produced and links to the original source. You will also see details of how the genes were annotated.
The current genome assembly for human is GRCh38. If you want to see the previous assembly, GRCh37, visit our dedicated site grch37.ensembl.org.
Ensembl Genomes
Homepage
Let’s take a look at the Ensembl Genomes homepage at ensemblgenomes.org.
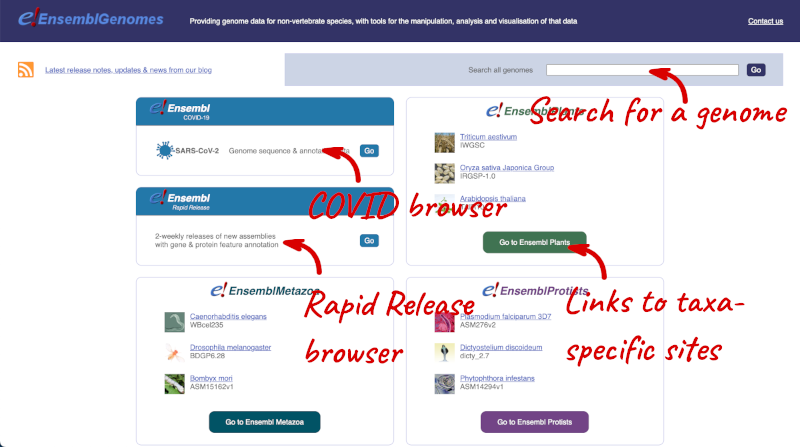
Click on the different taxa to see their homepages. Each one has a different colour-coding, but they are all structured in a similar format to the Ensembl main site.
You can navigate most of the taxa in the same way as you would with Ensembl.
Ensembl Bacteria
Ensembl Bacteria has a large number of genomes and has a slightly different method to the other Ensembl sites. Let’s look at it in more detail.
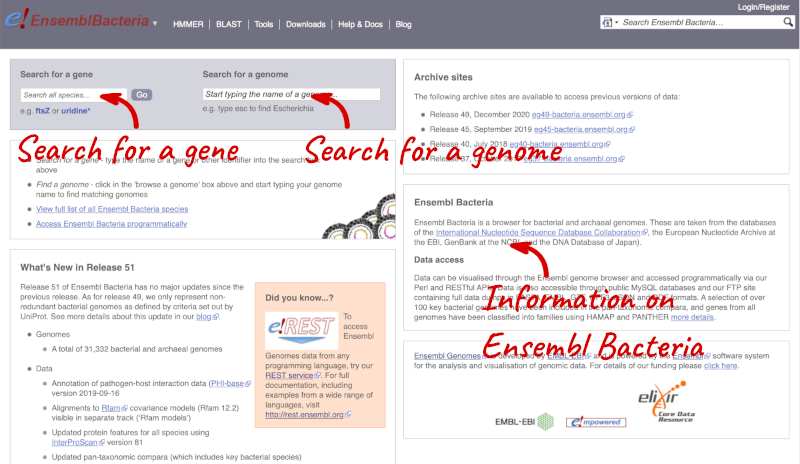
There’s no drop-down species list for bacteria as it would be hard to navigate with the number of species. You can click the View full list of all Ensembl Bacteria species link underneath the coloured search block. Search for your species of interest using the filter in the top right-hand corner of the table.
Alternatively, you can find a species by typing the species name into the Search for a genome search box at the top of the page. A drop-down list will appear with any species matching the name you entered.
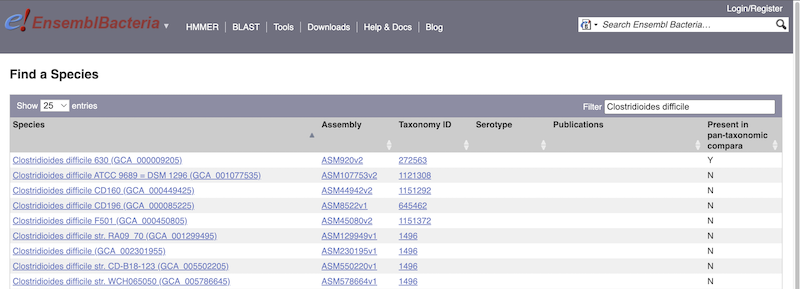
For example, to find a sub-strain of Clostridioides difficile start typing in the species name. Due to the auto-complete, you’ll see useful results as soon as you get to Clostridio.
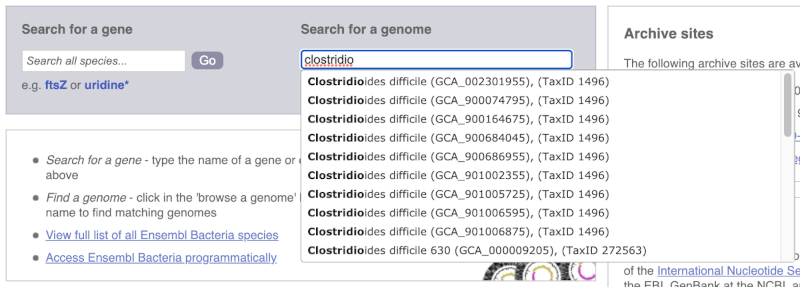
The drop down contains various strains of C. difficile. Let’s choose C. difficile 630. This will take us to another species information page, where we can explore various features.
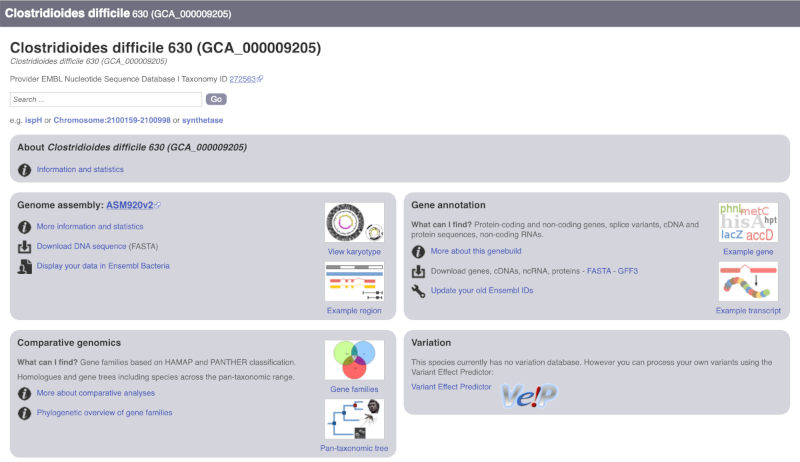
Unlike the Homo sapiens species information page, there is no prose description of the genome or gene annotation, as these pages were generated automatically.
Ensembl Rapid Release
Our newest genomes, such as those coming from the Darwin Tree of Life, are available rapid.ensembl.org with limited annotation.
Panda species
Go to Ensembl and find the following information:
-
What is the name of the genome assembly for Panda?
-
How long is the Panda genome (in bp)? How many coding genes have been annotated?
-
Select Giant panda from the drop down species list, or click on View full list of all Ensembl species, then choose Giant panda from the list.
The assembly is ASM200744v2 or GCA_002007445.2. -
Click on More information and statistics. Statistics are shown in the tables on the left.
The length of the genome is 2,444,060,653 bp.
There are 20,857 coding genes.
Available zebrafish assemblies
What previous assemblies are available for zebrafish?
Click on Zebrafish on the front page of Ensembl to go to the species homepage. Under Other assemblies three previous assembly names and the releases you can find them in are listed.
Assembly GRCz10 is available in the archived release 80, Zv9 in 77 and Zv8 in 54.
Solanum genus
Go to Ensembl Plants and answer the following questions:
-
How many genomes of the genus Solanum are there in Ensembl Plants?
-
When was the current Solanum lycopersicum genome assembly last revised?
- On the homepage, click on View full list of all Ensembl Plants species underneath the coloured search block. Type Solanum into the filter box in the top left-hand corner of the table.
There are three Solanum genomes: Solanum lycopersicum (tomato), and Solanum tuberosum RH89-039-16 and Solanum tuberosum (both potato).
- Click on S. lycopersicum, then on More information and statistics.
The genome was revised in April 2018.
Mosquito species
-
Go to Ensembl Metazoa. How many genomes relating to the genus Anopheles are there in Ensembl Metazoa?
-
When was the current Anopheles gambiae genome assembly last revised?
- Go to metazoa.ensembl.org. Open the drop-down list or click on View full list of all Ensembl Metazoa species. In a latin binomial species name, the first word represents the genus. Type Anopheles into the filter box in the top left to find all genomes with this word in the binomial.
There are 22 Anopheles genomes (some species are represented by more than one genome).
- Click on Anopheles gambiae (African malaria mosquito, PEST), and then on More information and statistics.
The assembly hosted is AgamP4 (INSDC Assembly GCA_000005575.1) which was revised in Feb 2006.
Finding a genome in Ensembl Bacteria
Mycobacterium tuberculosis H37Ra str. ATCC25177 is a clinical strain.
Go to Ensembl Bacteria and find the species M. tuberculosis H37Ra str. ATCC25177. How many coding genes does it have?
In the Ensesmbl Bacteria homepage, start to type H37Ra into the Search for a genome search box (you can find this in the coloured block at the top of the homepage). It will auto-complete, allowing you to select M. tuberculosis H37Ra str. ATCC25177 from the drop-down list. Click on More information and statistics.
M. tuberculosis H37Ra str. ATCC25177 has 4,080 coding and 47 non-coding genes.
Exploring genomic regions
Start at the Ensembl front page, ensembl.org. You can search for a region by typing it into a search box, but you have to specify the species.
To bypass the text search, you need to input your region coordinates in the correct format, which is chromosome, colon, start coordinate, dash, end coordinate, with no spaces for example: human 4:122868000-122946000. Type (or copy and paste) these coordinates into either search box.
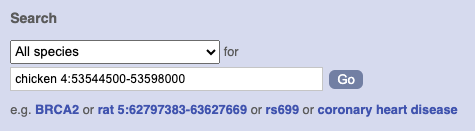
Press Enter or click Go to jump directly to the Region in detail Page.
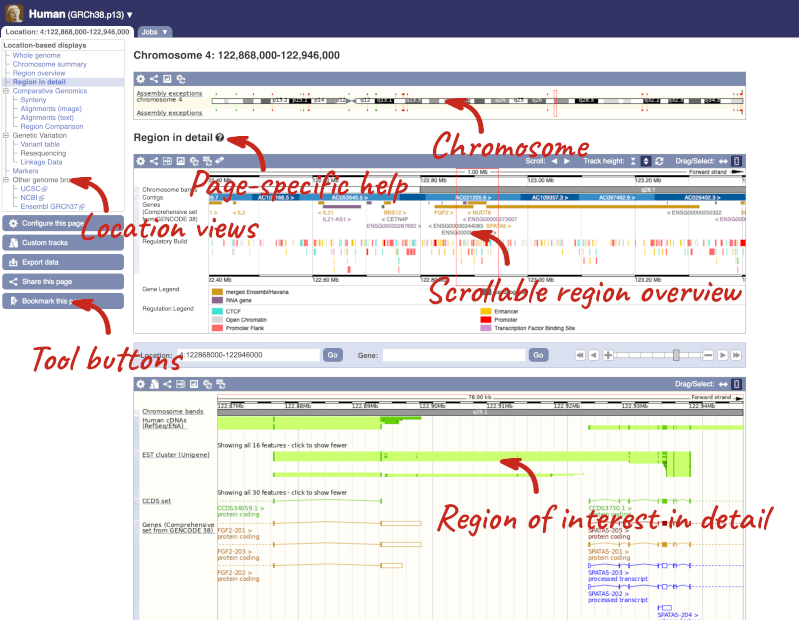
Click on the  button to view page-specific help. The help pages provide text, labelled images and, in some cases, help videos to describe what you can see on the page and how to interact with it.
button to view page-specific help. The help pages provide text, labelled images and, in some cases, help videos to describe what you can see on the page and how to interact with it.
The Region in detail page is made up of three images, let’s look at each one in detail.
The first image shows the chromosome:
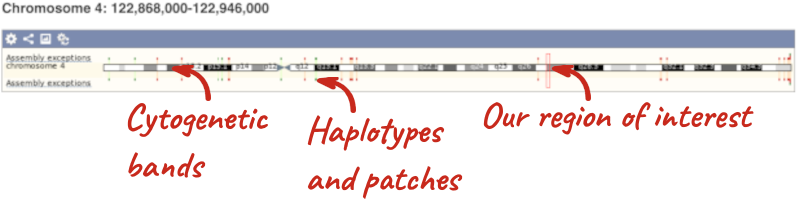
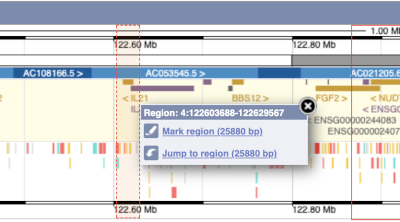
The region we’re looking at is highlighted on the chromosome. You can jump to a different region by dragging out a box in this image. Drag out a box on the chromosome, a pop-up menu will appear.
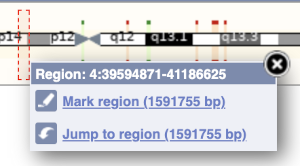
If you wanted to move to the region, you could click on Jump to region (### bp). If you wanted to highlight it, click on Mark region (###bp). For now, we’ll close the pop-up by clicking on the X on the corner.
The second image shows a 1Mb region around our selected region. This is always 1Mb in human, but the fixed size of this view varies between species. This view allows you to scroll back and forth along the chromosome.
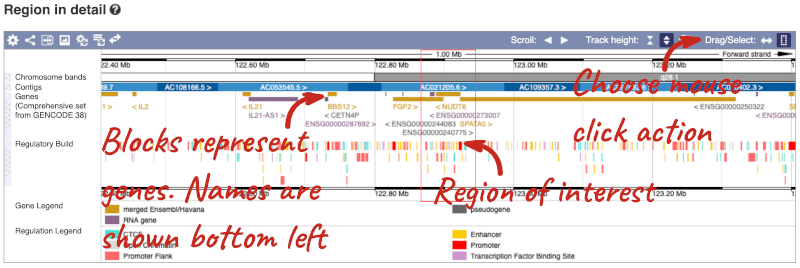
You can also drag out and jump to or mark a region.
Click on the X to close the pop-up menu.
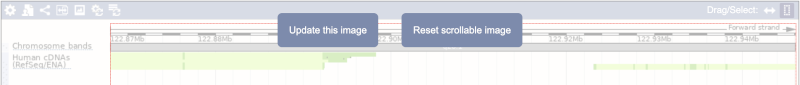
Click on the Drag/Select button  to change the action of your mouse click. Now you can scroll along the chromosome by clicking and dragging within the image. As you do this you’ll see the image below grey out and two blue buttons appear. Clicking on Update this image would jump the lower image to the region central to the scrollable image. We want to go back to where we started, so we’ll click on Reset scrollable image.
to change the action of your mouse click. Now you can scroll along the chromosome by clicking and dragging within the image. As you do this you’ll see the image below grey out and two blue buttons appear. Clicking on Update this image would jump the lower image to the region central to the scrollable image. We want to go back to where we started, so we’ll click on Reset scrollable image.
The third image is a detailed, configurable view of the region.

Here you can see various tracks, which is what we call a data type that you can plot against the genome. Some tracks, such as the transcripts, can be on the forward or reverse strand. Forward stranded features are shown above the blue contig track that runs across the middle of the image, with reverse stranded features below the contig. Other tracks, such as variants, regulatory features or conserved regions, refer to both strands of the genome, and these are shown by default at the very top or very bottom of the view.
You can use click and drag to either navigate around the region or highlight regions of interest, Click on the Drag/Select option at the top or bottom right to switch mouse action. On Drag, you can click and drag left or right to move along the genome, the page will reload when you drop the mouse button. On Select you can drag out a box to highlight or zoom in on a region of interest.
With the tool set to Select, drag out a box around an exon and choose Mark region.
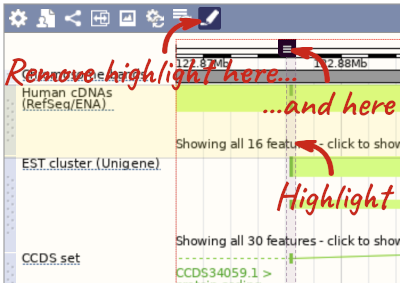
The highlight will remain in place if you zoom in and out or move around the region. This allows you to keep track of regions or features of interest.
We can edit what we see on this page by clicking on the blue Configure this page menu at the left.

This will open a menu that allows you to change the image.
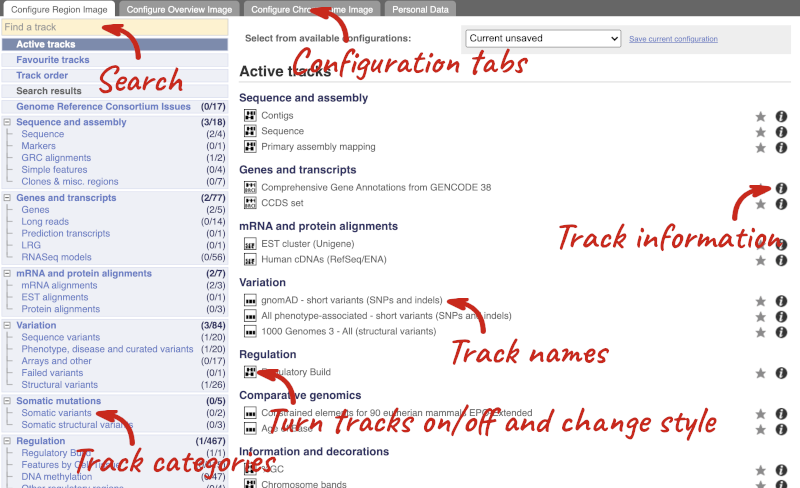
There are thousands of possible tracks that you can add. When you launch the view, you will see all the tracks that are currently turned on with their names on the left and an info icon on the right, which you can click on to expand the description of the track. Turn them on or off, or change the track style by clicking on the box next to the name. More details about the different track styles are in this FAQ: http://www.ensembl.org/Help/Faq?id=335.
You can find more tracks to add by either exploring the categories on the left, or using the Find a track option at the top left. Type in a word or phrase to find tracks with it in the track name or description.
Let’s add some tracks to this image. Add:
- Proteins (mammal) from UniProt – Labels
- 1000 Genomes - All - short variants (SNPs and indels) – Normal
Now click on the tick in the top left hand to save and close the menu. Alternatively, click anywhere outside of the menu. We can now see the tracks in the image. The proteins track is stranded, so you will see two tracks, one above and one below the contig, representing the proteins mapped to the forward and reverse strands respectively. The variants track is not stranded, so is found near the bottom of the image.
If the track is not giving you can information you need, you can easily change the way the tracks appear by hovering over the track name then the cog wheel to open a menu. To make it easier to compare information between tracks, such as spotting overlaps, you can move tracks around by clicking and dragging on the bar to the left of the track name.

Now that you’ve got the view how you want it, you might like to show something you’ve found to a colleague or collaborator. Click on the Share this page button to generate a link. Email the link to someone else, so that they can see the same view as you, including all the tracks you’ve added. These links contain the Ensembl release number, so if a new release or even assembly comes out, your link will just take you to the archive site for the release it was made on.
To return this to the default view, go to Configure this page and select Reset configuration at the bottom of the menu.
Exploring a genomic region in human
Go to Ensembl.
-
Go to the region from 32,264,000 to 32,492,000 bp on human chromosome 13. On which cytogenetic band is this region located? How many contigs make up this portion of the assembly (contigs are contiguous stretches of DNA sequence that have been assembled solely based on direct sequencing information)?
-
Zoom in on the BRCA2 gene.
-
Configure this page to turn on the LTR (repeat) track in this view. What tool was used to annotate the LTRs according to the track information? How many LTRs can you see within the BRCA2 gene? Do any overlap exons?
-
Create a Share link for this display. Email it to your neighbour. Open the link they sent you and compare. If there are differences, can you work out why?
-
Export the genomic sequence of the region you are looking at in FASTA format.
-
Turn off all tracks you added to the Region in detail page.
- Go to the Ensembl homepage, select Human from the Species drop-down list and type
13:32264000-32492000in the text box (alternatively leave the Search drop-down list as it is and type13:32264000-324920000in the text box). Click Go.This genomic region is located on cytogenetic band q13.1. It is made up of three contigs, indicated by the alternating light and dark blue coloured bars in the Contigs track.
-
Draw with your mouse a box encompassing the BRCA2 transcripts. Click on Jump to region in the pop-up menu.
- Click Configure this page in the side menu (or on the cog wheel icon in the top left hand side of the bottom image). Go into Repeats in the left-hand menu then select LTR. Click on the (i) button to find out more information.
Repeat Masker was used to annotate LTRs onto the genome.
Save and close the new configuration by clicking on ✓ (or anywhere outside the pop-up window). There are ten LTRs overlapping BRCA2, none of them overlap exons. -
Click Share this page in the side menu. Copy the URL. Get your neighbour’s email address and compose an email to them, paste the link in and send the message. When you receive the link from them, open the email and click on your link. You should be able to view the page with the new configuration and data tracks they have added to in the Location tab. You might see differences where they specified a slightly different region to you, or where they have added different tracks.
Here is the Share link from the video answer: https://may2021.archive.ensembl.org/Homo_sapiens/Share/71a173bba78f0dbe03e48d3240424943?redirect=no;mobileredirect=no
-
Click Export data in the side menu. Leave the default parameters as they are (FASTA sequence should already be selected). Click Next>. Click on Text. Note that the sequence has a header that provides information about the genome assembly (GRCh38), the chromosome, the start and end coordinates and the strand. For example:
>13_dna:chromosome_chromosome:GRCh38:13:32311910:32405865:1 - Click Configure this page in the side menu. Click Reset configuration. Click ✓.
Exploring a genomic region in mouse
Go to the Ensembl homepage.
-
Go to the region from 150,320,000 to 150,540,000 bp on mouse chromosome 5. How many contigs make up this portion of the assembly (contigs are contiguous stretches of DNA sequence that have been assembled solely based on direct sequencing information)?
-
Zoom in on the Brca2 gene.
-
Configure this page to turn on the LTR (repeat) track in this view. What tool was used to annotate the LTRs according to the track information? How many LTRs can you see within the Brca2 gene? Do any overlap exons?
-
Create a Share link for this display. Email it to your neighbour. Open the link they sent you and compare. If there are differences, can you work out why?
-
Export the genomic sequence of the region you are looking at in FASTA format.
-
Turn off all tracks you added to the Region in detail page.
- Select Mouse from the Species search list and type
5:150320000-150540000in the text box (or alternatively leave the Search drop-down list like it is and typemouse 5:150320000-150540000in the text box). Click Go.It is made up of five contigs, indicated by the alternating light and dark blue coloured bars in the Contigs track. Note the tiny contig, AEKQ02165236.1, which splits AC084217.7 in two.
-
Draw with your mouse a box encompassing the Brca2 transcripts. Click on Jump to region in the pop-up menu.
- Click Configure this page in the side menu (or on the cog wheel icon in the top left hand side of the bottom image). Go to Repeats in the left-hand menu then select LTRs (Repeats (Mouse)). Click on the (i) button to find out more information.
Repeat Masker was used to annotate LTRs onto the genome.
Save and close the new configuration by clicking on ✓ (or anywhere outside the pop-up window).
There are seven LTRs overlapping Brca2, none of them overlap exons.
-
Click Share this page in the side menu. Select the link and copy. Get your neighbour’s email address and compose an email to them, paste the link in and send the message. When you receive the link from them, open the email and click on your link. You should be able to view the page with the new configuration and data tracks they have added to in the Location tab. You might see differences where they specified a slightly different region to you, or where they have added different tracks.
-
Click Export data in the side menu. Leave the default parameters as they are. Click Next>. Click on Text.
- Click Configure this page in the side menu. Click Reset configuration. Click ✓.
Exploring a genomic region in Oryza sativa Japonica (rice)
Go to the Ensembl Plants homepage and do the following:
-
Go to the region between 405000 and 453000 on chromosome 1 in Oryza sativa Japonica.
-
Turn on the AGILENT:G2519F-015241 microarray track. Are there any oligo probes that map to this region?
-
Highlight the region around any reverse strand probes you can see. Do they map to any Ensembl transcripts?
-
Go to the Ensembl Plants homepage. Select Oryza sativa Japonica from the Species drop-down list and type
1:405000-453000. Click Go. - Click on Configure this page to open the menu. You can find the AGILENT:G2519F-015241 track under Oligo probes in the left-hand menu, or by using the Find a track box at the top right. Turn on the track as Normal then save and close the menu. As the AGILENT:G2519F-015241 track is stranded, it appears at the top and bottom of the view.
There are 5 probes mapped to this region on the positive strand and one probe on the reverse strand.
- Drag a box around the reverse strand probe then click on Mark region to highlight.
The highlighted region maps to two transcripts: Os01t0107900-02 and Os01t0107900-01
Exploring a region in Coprinopsis cinerea okayama
Go to Ensembl Fungi. Let’s try to find some information about the region from 1,400,000 to 1,425,000 in chromosome 7 in Coprinopsis cinerea okayama:
-
How many complete genes are found in this region? How many on the forward and how many on the reverse strand?
-
Zoom in on the largest gene EFI27358. How many exons does this gene have?
-
Export the genomic sequence in FASTA format for this region.
- In the Ensembl Fungi homepage, select Coprinopsis cinerea okayama from the Species search drop-down. Enter
7:1400000-1425000in the Search bar and click Go. This will send you to the Location tab. Your region of interest is indicated by a red rectangle in the 50kb view. Look at the Genes track: each block represents a different gene. Count the number of complete genes within the rectangle.There are 7 complete genes in the region.
- Look at the Region in detail view (the most detailed view at the bottom of the page). You can zoom into a region by clicking and dragging your mouse (you can change your mouse action in the top right-hand corner of the view under **Drag/Select) and selecting Jump to region in the pop-up menu. Count the number of blocks you can see for EFI27358.
The EFI27358 gene has 23 exons.
Click on the transcript ID CZT99117 in the transcript table.
It has 4 exons.
- We want to export the genomic sequence for our original region (not just the EFI27358 gene). You can reset the view by entering
7:1400000-1425000in the Location bar above the Region in detail view or hitting the Back button on your internet browser. Click on Export data in the left-hand panel. In the pop-up menu, select FASTA from the drop-down and click Next >. You can export the sequence as is (text) or as a compressed file (.gz).If you choose to download the sequence as text, your browser might open the FASTA file in a new tab. In this case, just right-click on any white space and select Save As… from the menu.
Exploring a genomic region in Salmonella enterica
Go to Ensembl Bacteria and do the following:
-
Search for the Salmonella enterica subsp. enterica serovar Typhi str. Ty2 (GCA_000007545) (Hint: type
Tyinto the Search for a genome box). -
Go to the region Chromosome:2000605-2009742.
-
How many genes are annotated in this region? How many are on the forward strand? How many are on the reverse strand?
-
Go to the Ensembl Bacteria homepage. Type
Ty2into the Search for a genome box. Click on the auto-completed genome name to navigate to the species information page. -
Type
Chromosome:2000605-2009742into the search box. Click Go. -
There are 8 genes annotated in this region, all on the reverse strand.
Genes and transcripts
You can find out lots of information about Ensembl genes and transcripts using the browser. If you’re already looking at a region view, you can click on any transcript and a pop-up menu will appear, allowing you to jump directly to that gene or transcript.
Alternatively, you can find a gene by searching for it. You can search for gene names or identifiers, and also phenotypes or functions that might be associated with the genes.
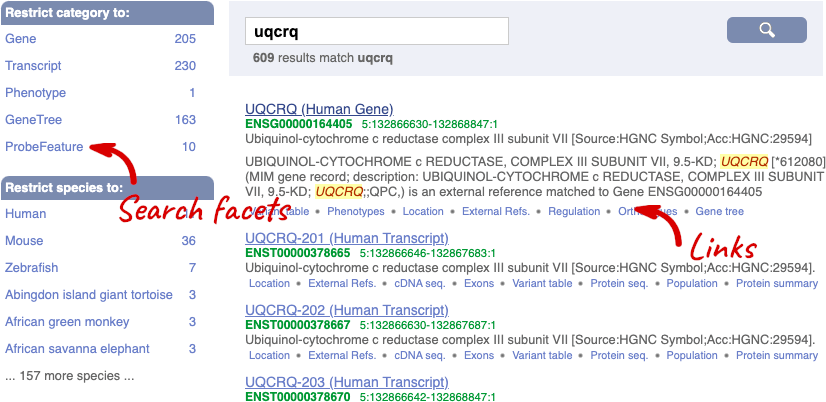
We’re going to look at the human UQCRQ gene. From ensembl.org, type UQCRQ into the search bar and click the Go button. You will get a list of hits with the human gene at the top.
Where you search for something without specifying the species, or where the ID is not restricted to a single species, the most popular species will appear first, in this case, human, mouse and zebrafish appear first. You can restrict your query to species or features of interest using the options on the left.
The gene tab
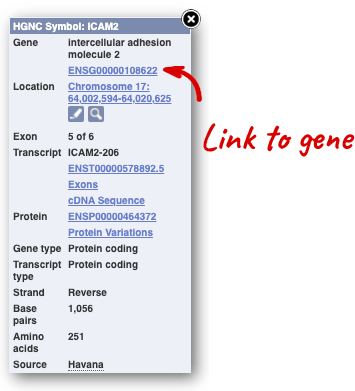
Click on the gene name or Ensembl ID. The Gene tab should open:
This page summarises the gene, including its location, name and equivalents in other databases. At the bottom of the page, a graphic shows a region view with the transcripts. We can see exons shown as blocks with introns as lines linking them together. Coding exons are filled, whereas non-coding exons are empty. We can also see the overlapping and neighbouring genes and other genomic features.
There are different tabs for different types of features, such as genes, transcripts or variants. These appear side-by-side across the blue bar, allowing you to jump back and forth between features of interest. Each tab has its own navigation column down the left hand side of the page, listing all the things you can see for this feature.
Let’s walk through this menu for the gene tab. How can we view the genomic sequence? Click Sequence at the left of the page.
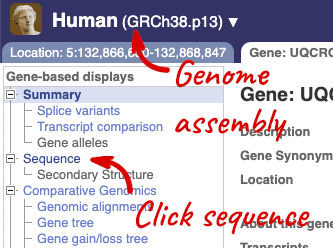
The sequence is shown in FASTA format. The FASTA header contains the genome assembly, chromosome, coordinates and strand (1 or -1) – this gene is on the positive strand.
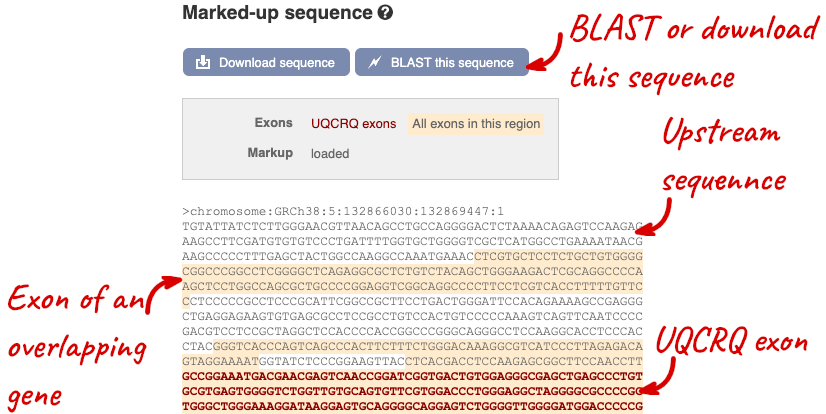
Exons are highlighted within the genomic sequence, both exons of our gene of interest and any neighbouring or overlapping gene. By default, 600 bases are shown up and downstream of the gene. We can make changes to how this sequence appears with the blue Configure this page button found at the left. This allows us to change the flanking regions, add variants, add line numbering and more. Click on it now.
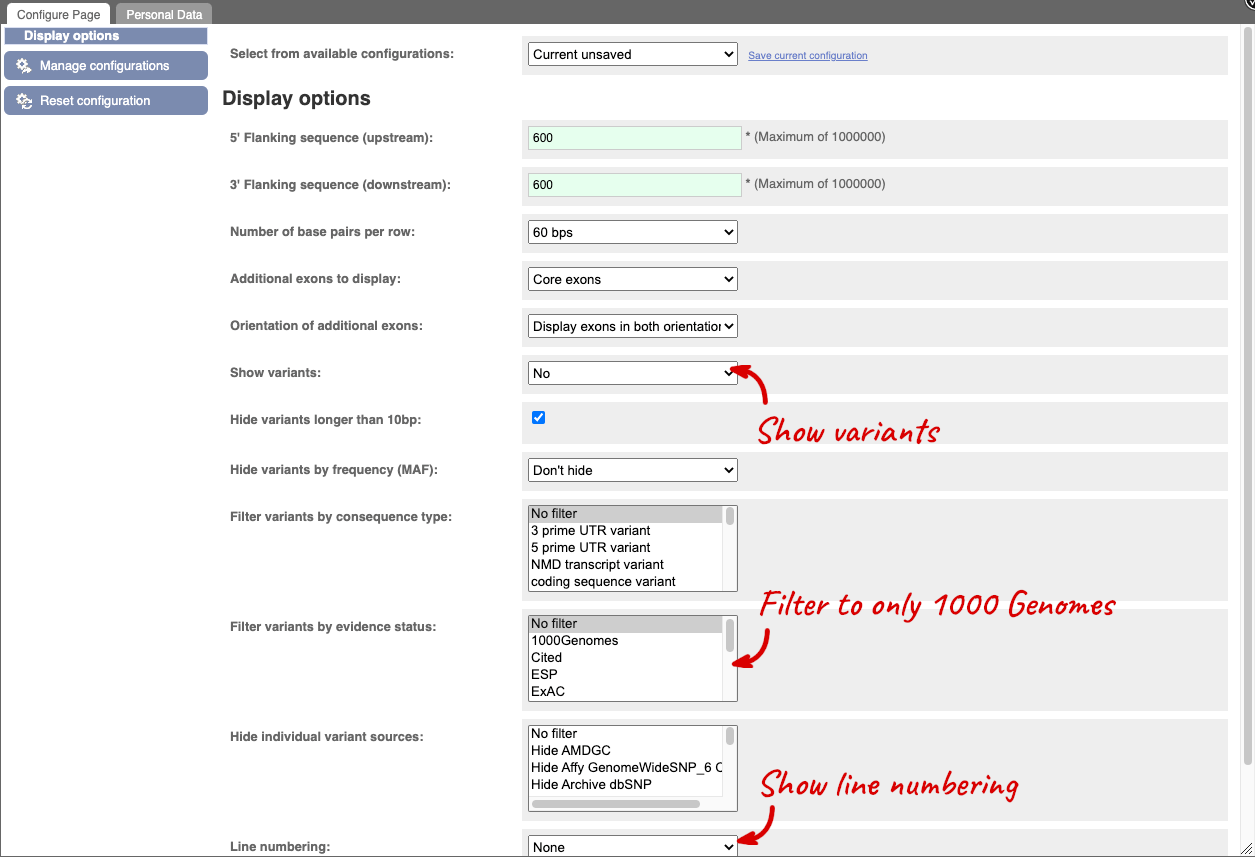
Once you have selected changes (in this example, Show variants, 1000 Genomes variants and Line numbering) click at the top right.
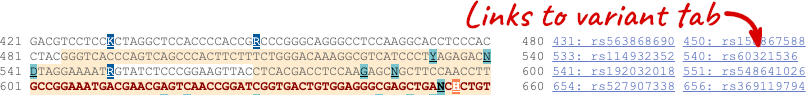
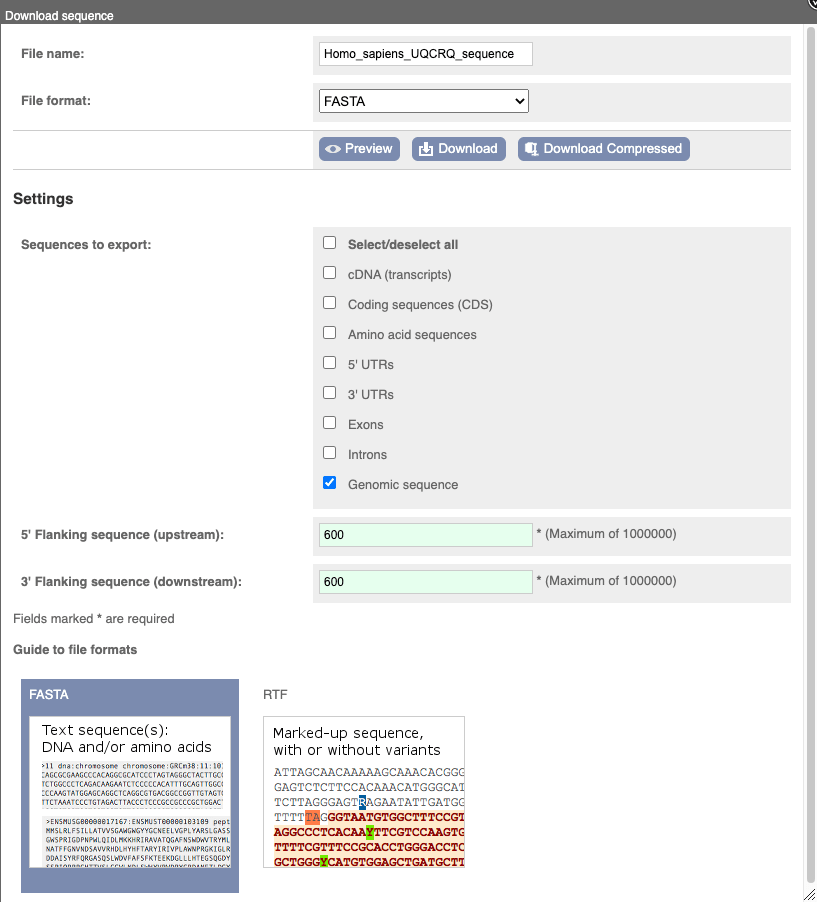
You can download this sequence by clicking in the Download sequence button above the sequence:

This will open a dialogue box that allows you to pick between plain FASTA sequence, or sequence in RTF, which includes all the coloured annotations and can be opened in a word processor. If you want run a sequence analysis tool, download as FASTA sequence, whereas if you want to analyse the sequence visually, RTF is best for this. This button is available for all sequence views.
To find out what the protein does, have a look at GO terms from the Gene Ontology consortium. There are three pages of GO terms, representing the three divisions in GO: Biological process (what the protein does), Cellular component (where the protein is) and Molecular function (how it does it). Click on GO: Biological process to see an example of the GO pages.

Here you can see the functions that have been associated with the gene. There are three-letter codes that indicate how the association was made, as well as links to the specific transcript they are linked to.
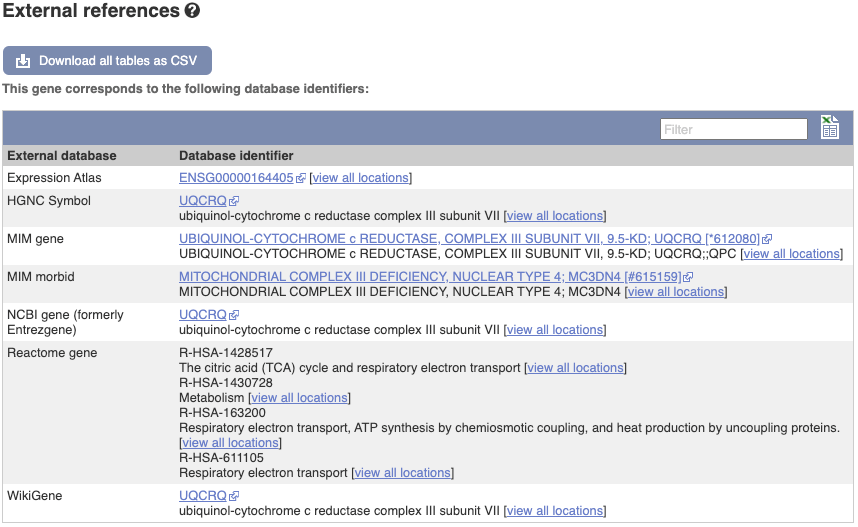
We also have links out to other databases which have information about our genes and may focus on other topics that we don’t cover, like Gene Expression Atlas or OMIM. Go up the left-hand menu to External references:
Demo: The transcript tab
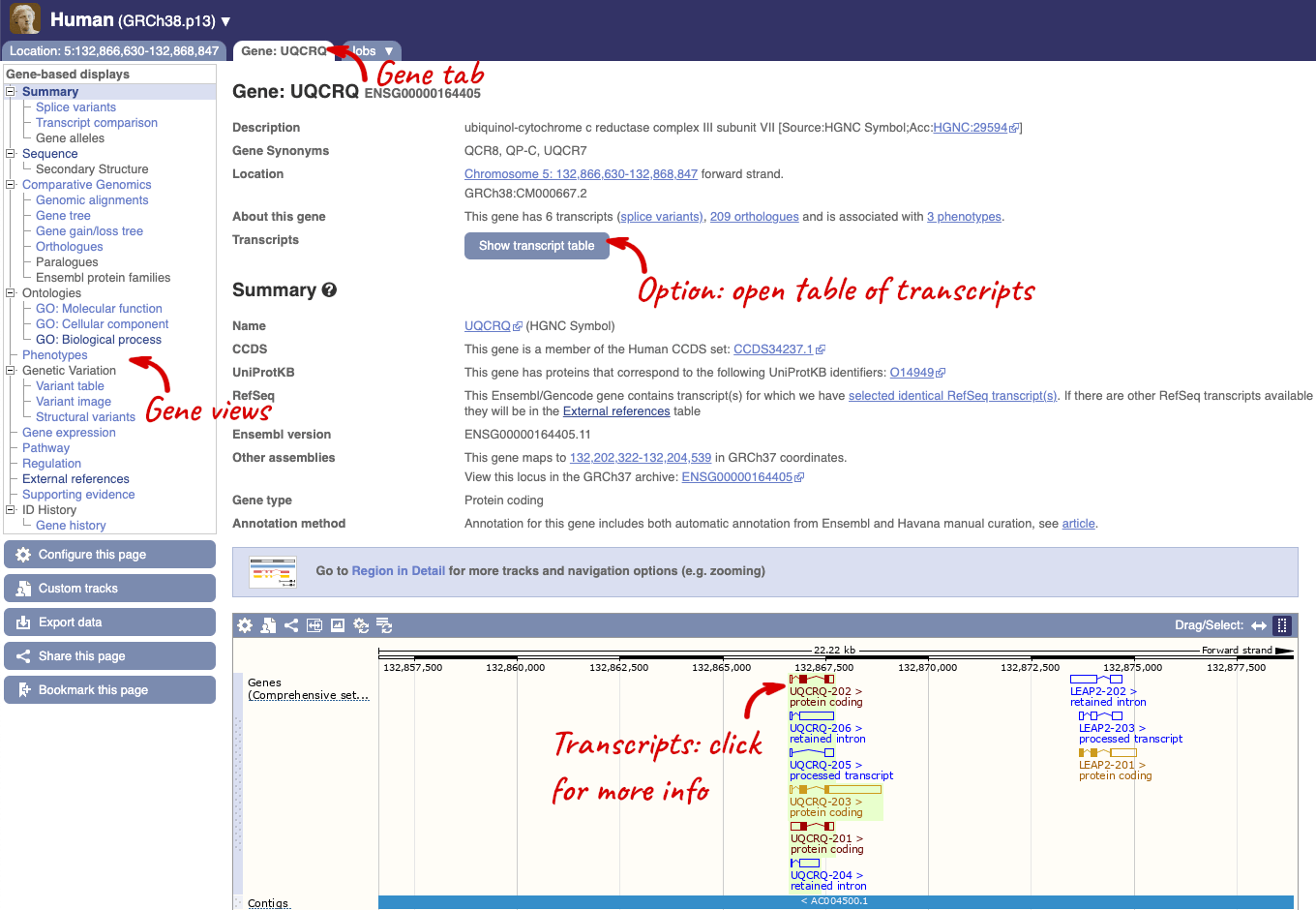
We’re now going to explore the different transcripts of UQCRQ. Click on Show transcript table at the top.
![]()
![]()
Here we can see a list of all the transcripts of UQCRQ with their identifiers, lengths, biotypes and flags to help you decide which ones to look at.
If we were to only choose one transcript to analyse, we would choose UQCRQ-203 because it is the MANE Select and Ensembl Canonical. This means it is both 100% identical to the RefSeq transcript NM_014402.5 and both Ensembl and NCBI agree that it is the most biologically important transcript.
Click on the ID, ENST00000378670.8.
You are now in the Transcript tab for UQCRQ-203. We can still see the gene tab so we can easily jump back. The left hand navigation column provides several options for the transcript UQCRQ-203 - many of these are similar to the options you see in the gene tab, but not all of them. If you can’t find the thing you’re looking for, often the solution is to switch tabs.
Click on the Exons link. This page is useful for designing RT-PCR primers because you can see the sequences of the different exons and their lengths.
![]()
You may want to change the display (for example, to show more flanking sequence, or to show full introns). In order to do so click on Configure this page and change the display options accordingly.
Now click on the cDNA link to see the spliced transcript sequence with the amino acid sequence. This page is useful for mapping between the RNA and protein sequences, particularly genetic variants.
![]()
UnTranslated Regions (UTRs) are highlighted in dark yellow, codons are highlighted in light yellow, and exon sequence is shown in black or blue letters to show exon divides. Sequence variants are represented by highlighted nucleotides and clickable IUPAC codes are above the sequence.
Next, follow the General identifiers link at the left. Just like the External References page in the gene tab, this page shows links out to other databases such as RefSeq, UniProtKB, PDBe and others, this time linked to the transcript or protein product, rather than the gene.
![]()
If you’re interested in protein domains, you could click on Protein summary to view domains from Pfam, PROSITE, Superfamily, InterPro, and more. These are all plotted against the transcript sequence, with the exons shown in alternating shades of purple at the top of the page. Alternatively, you can go to Domains & features to see a table of the same information.
![]()
![]()
You can also see the structure of the protein from the PDB by clicking on PDB 3D Protein model.
![]()
This uses LiteMol to show a 3D protein. You can use all the normal controls that you would use with LiteMol, plus plot Ensembl features like Exons and variants onto the structure using the options on the right. We allow you to see the top ten PDB models for this protein, based on coverage and quality scores, you can choose which at the top of the viewer.
Exploring the MYH9 gene in human
- In Ensembl, find the human MYH9 (myosin, heavy chain 9, non-muscle) gene and open the Gene tab.
- On which chromosome and which strand of the genome is this gene located?
- How many transcripts (splice variants) are there and how many are protein coding?
- What is the longest protein-coding transcript, and how long is the protein it encodes?
- Which transcript would you take forward for further study?
-
Click on Phenotypes at the left side of the page. Are there any diseases associated with this gene, according to Mendelian Inheritance in Man (MIM)?
-
What are some functions of MYH9 according to the Gene Ontology (GO) consortium? Have a look at the GO: Biological process pages for this gene.
- In the transcript table, click on the transcript ID for MYH9-201, and go to the Transcript tab.
- How many exons does it have?
- Are any of the exons completely or partially untranslated?
- Is there an associated sequence in UniProtKB/Swiss-Prot? Have a look at the General identifiers for this transcript.
- Are there microarray (oligo) probes that can be used to monitor ENST00000216181 expression?
- Select Human from the Species drop-down list and type
MYH9. Click Go. Click on MYH9 (Human Gene) in the search results which will send you to the Gene tab.- The gene is located on chromosome 22 on the reverse strand.
- Ensembl has 23 transcripts annotated for this gene, of which 6 are protein-coding.
- The longest protein-coding transcript is MYH9-215 and it codes for a protein that is 1,981 amino acids long.
- MYH9-201 is the transcript I would take forward for further study, as it is the MANE Select transcript (for a description, mouse-over the MANE Select flag in the transcript table).
- Click on Phenotypes in the left-hand panel to see the associated phenotypes. There is a large table of phenotypes. To see only the ones from MIM, type
MIMinto the filter box at the top right-hand corner of the table.These are some of the phenotypes associated with MYH9 according to MIM: Deafness, Autosomal dominant 17 and Macrothrombocytopenia and granulocyte inclusions with or without nephritis or sensorineural hearing loss. You can click on the records for more information.
-
The Gene Ontology project maps terms to a protein in three classes: biological process, cellular component, and molecular function. Click on GO: Biological process on the left-hand panel. Angiogenesis, cell adhesion, and protein transport are some of the roles associated with MYH9. All GO terms are associated with a single transcript: ENST00000216181.
- Click on ENST00000216181.11 in the transcript table. You should now be on the Transcript tab.
- It has 41 exons, shown in the Transcript summary.
Click on the Exons link in the left-hand panel.
- Exon 1 is completely untranslated, and exons 2 and 41 are partially untranslated (UTR sequence is shown in orange). You can also see this in the cDNA view if you click on the cDNA link in the left side menu.
Click on General identifiers in the left-hand panel.
- P35579.254 from UniProt/Swiss-Prot matches the translation of the Ensembl transcript. Click on P35579.254 to go to UniProtKB, or click align for the alignment.
- Click on Oligo probes in the left-hand panel.
Probesets from Affymetrix, Agilent, Codelink, Illumina, and Phalanx OneArray match to this transcript sequence. Expression analysis with any of these probesets would reveal information about the transcript. Hint: this information can sometimes be found in the [ArrayExpress Atlas] (https://www.ebi.ac.uk/biostudies/arrayexpress).
Finding a gene associated with a phenotype
Phenylketonuria is a genetic disorder caused by an inability to metabolise phenylalanine in any body tissue. This results in an accumulation of phenylalanine causing seizures and intellectual disability.
(a) Search for phenylketonuria from the Ensembl homepage and narrow down your search to only genes. What gene is associated with this disorder?
(b) How many protein coding transcripts does this gene have? View all of these in the transcript comparison view.
(c) What is the MIM gene identifier for this gene?
(d) Go to the MANE Select transcript and look at its 3D structure. In the model 2pah, how many protein molecules can you see?
(a) Start at the Ensembl homepage (http://www.ensembl.org).
Type phenylketonuria into the search box then click Go. Choose Gene from the left hand menu.
The gene associated with this disorder is PAH, phenylalanine hydroxylase, ENSG00000171759.
(b) If the transcript table is hidden, click on Show transcript table to see it.
There are six protein coding transcripts.
Click on Transcript comparison in the left hand menu. Click on Select transcripts. Either select all the transcripts labelled protein coding one-by-one, or click on the drop down and select Protein coding. Close the menu.
(c) Click on External references.
The MIM gene ID is 612349.
(d) Open the transcript table and click on the ID for the MANE Select: ENST00000553106.6. Go to PDB 3D protein model in the left-hand menu.
The model 2pah is shown by default. It has two protein molecules in it. You may need to rotate the model to see this clearly.
Exploring the Dpp6 gene in mouse
Genetic variation in the dipeptidylpeptidase 6 Gene (DPP6) in humans has previously been strongly associated with amyotrophic lateral sclerosis (ALS), a lethal disorder caused by progressive degeneration of motor neurons in the brain.
-
Go to the Ensembl homepage, search for the Dpp6 gene in mouse and click on the transcript ID ENSMUST00000071500 to open the transcript tab. How many exons make up this transcript?
-
Click on Exons to display the exon sequences of the transcript. Which exon contains the translation start? What is the exon ID of the largest exon? What is the start and end phase of exon 2?
-
Go to the Protein summary. How many protein domains or features fall within the second exon? What is the Pfam protein domain at the C-terminus of the protein and how many exons does it fall into? Which amino acid positions does the domain above cover?
-
Go to Domains and features. Which domains are associated with Pfam? How many genes in the mouse genome have the IPR002469 domain? What chromosomes are these genes found on?
- Select Mouse from the Species search drop-down and type
Dpp6and click Go. Click on Dpp6-201 (Mouse Transcript, Strain: reference (CL57BL6)) in the results.ENSMUST00000071500.13 consists of 26 exons.
- Click on Exons in the left-hand panel. The translation start is found in the first exon (ENSMUSE00000725552), shown in dark blue text.
The largest exon is the final exon (856 bp), which has the exon ID ENSMUSE00000773588. Exon 2 has a start and end phase of 0 and 1 respectively, which means that the codon at the start of the exon starts at the first nucleotide and the codon at the end of the exon ends at nucleotide 2. Notice that the end phase of each exon is the same as the start phase of the next exon.
- Click on Protein summary in the menu on the left hand side of the page. Alternating exons are shown on the protein as different shades of purple.
There are two predicted protein domains that fall within the second exon: a transmembrane helix and low complexity peptide sequence (Seg). You can click on the track names to find a description.
Click on a domain or feature to view further information.
The C-terminal Pfam domain is Peptidase_S9 (PF00326), which spans or partially spans seven exons, covering amino acid positions 582-787.
- Click on Domains & features.
Looking at the domains table you should notice that there are two domains associated with Pfam: PF00326 and PF00930.
Click on Display all genes with this domain next to IPR002469. This should now display the genes that have the IPR002469 domain located on the karyotype and as a table.
6 genes have this domain and they are found on chromosomes 1, 2, 5, 9 and 17.
Exploring the CCD7 gene in Arabidopsis thaliana
-
Find the Arabidopsis thaliana CCD7 gene on Ensembl Plants. On which chromosome and which strand of the genome is this gene located?
-
Where in the cell is the CCD7 protein located?
-
What is the source of the assigned gene name?
-
How many transcripts does it have? How long is its longest transcript (in bp)? How long is the protein it encodes? How many exons does it have? Are any of the exons completely or partially untranslated?
- Go to the Ensembl Plants homepage (http://plants.ensembl.org/). Select A. thaliana from the species list and type
CCD7in the search box. Click Go and click on the gene ID AT2G44990. You can find the strand orientation and the location under Summary in the Gene tab.The A. thaliana CCD7 gene is located on chromosome 2 on the forward strand.
- Click on GO: Cellular component in the left-hand panel.
The protein is located in the chloroplast and plastid.
- Click on Summary in the side menu.
The gene name is assigned and imported from NCBI gene (formerly Entrezgene).
- Click on Show transcript table.
There are 3 transcripts. The longest one is 2005 bp and the length of the encoded protein is 622 amino acids.
Click on the transcript ID AT2G44990.3 in the transcript table. You can find the number of exons in under in the summary information at the top of the page.
It has 6 exons.
Click on Sequence: Exons in the left-hand panel.
The first and last exons are partially untranslated (sequence shown in orange). This can also been seen from the fact that in the transcript diagrams on the Gene Summary and Transcript Summary pages the boxes representing the first and last exon are partially unfilled.
Exploring a bacterial gene in Clostridium sporogenes
Start in Ensembl Bacteria and select the Clostridium sporogenes (GCA_001444695) genome.
-
What is the gene name for the Glutamine synthetase gene?
-
Go to the transcript tab. How long is the transcript? How long is the protein?
-
What domains can be found in the protein product of this transcript? How many different domain prediction methods agree with each of these domains?
- From the Ensembl Bacteria homepage, select Clostridium sporogenes by beginning to write the species name and selecting the species from the auto-complete list. Type
Glutamine synthetaseand click on the gene ID ENSB:yZtlLO8Ti90y75J which will open the Summary display on the Gene tab..The gene name is
glnA. - Switch to the Transcript tab and go to the Summary display. You can find the length under Statistics underneath the transcript image.
The
glnAtranscript is 1,899 bp and the protein is 632 aa in length. - Click on either Protein Summary or Domains & features in the left hand menu to see graphically or as a table respectively.
6 protein domains were found. All of them predict a glutamine synthetase domain.
Exploring a gene in Escherichia coli
Start in Ensembl Bacteria and search for the Escherichia coli str. K-12 substr. MG1655 (GCA_000005845) genome.
-
What GO: biological process terms are associated with the Era gene?
-
How many different InterPro domains are found in the protein product of this gene?
-
What is the associated UniProt ID of the transcript?
Enter part of the name into the genome search box (e.g. MG1655) and then select the correct genome to go to the species information page.
- Enter
Erainto the search box and hit Go. Click the link in the first hit to go to the era gene page. From here, click GO: Biological process in the left-hand menu.There are three GO IDs: GO:0000028, GO:0006468, GO:0042274 and GO:0046777.
- Switch to the Transcript tab and go to Domains & features in the left-hand panel. Count the number of unique InterPro IDs in the table.
8 different InterPro domains are found in the protein product of Era.
- You can find the UniProt ID in the transcript table or under General identifiers in the left-hand panel.
The UniProtKB/Swiss-Prot ID is P06616.
Variation
In any of the sequence views shown in the Gene and Transcript tabs, you can view variants on the sequence. You can do this by clicking on Configure this page from any of these views.
Let’s take a look at the Gene sequence view for HBB in human. Search for HBB and go to the Sequence view.
If you can’t see variants marked on this view, click on Configure this page and select Show variants: Yes and show links. You may also wish to add a filter to the variants to allow them to load more quickly, we’ll add Filter variants by evidence status: 1000Genomes.
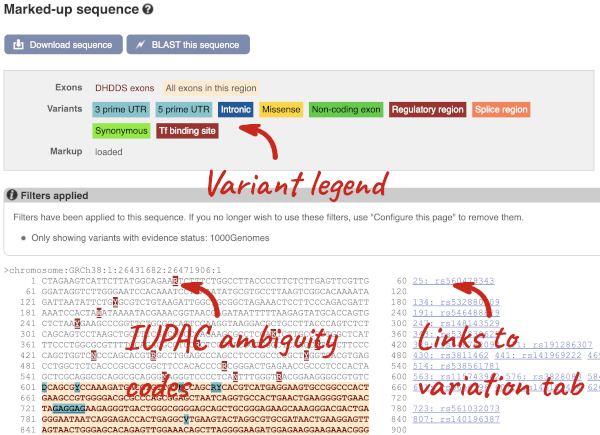
Find out more about a variant by clicking on it.
You can add variants to all other sequence views in the same way.
You can go to the Variation tab by clicking on the variant ID. For now, we’ll explore more ways of finding variants.
To view all the sequence variants in table form, click the Variant table link at the left of the gene tab.
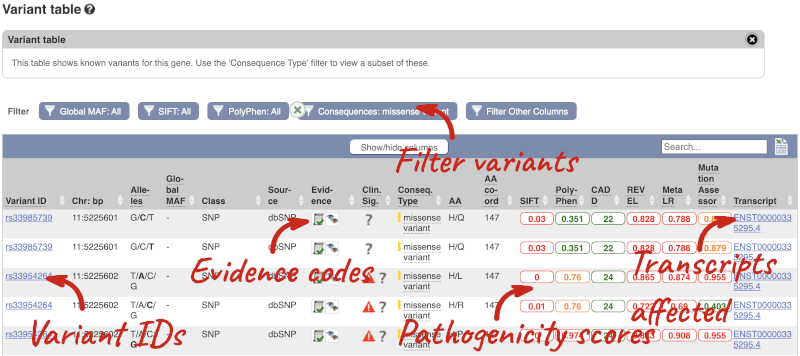
You can filter the table to only show the variants you’re interested in. For example, click on Consequences: All, then select the variant consequences you’re interested in. For display purposes, the table above has already been filtered to only show missense variants.
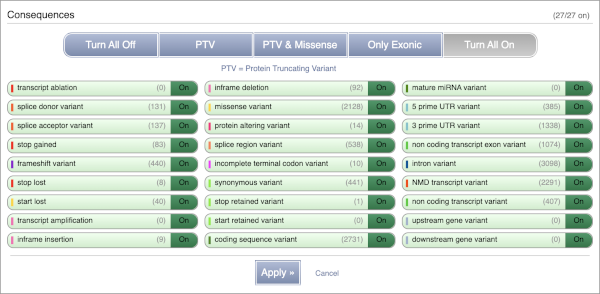
You can also filter by the different pathogenicity scores and MAF, or click on Filter other columns for filtering by other columns such as Evidence or Class.
The table contains lots of information about the variants. You can click on the IDs here to go to the Variation tab too.
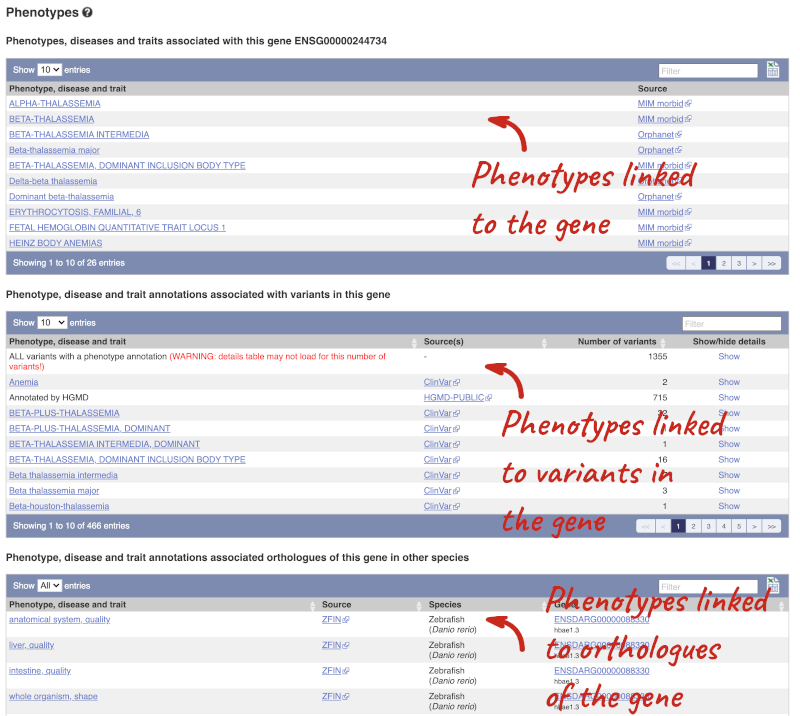
You can also see the phenotypes associated with a gene. Click on Phenotype in the left hand menu.
Open the transcript table and go to HBB-201 ENST00000335295, then click on Haplotypes in the left hand menu.
![]()
The Haplotypes view in the transcript tab shows you the actual protein and CDS sequences in 1000 Genomes individuals. This is possible because the 1000 Genomes study has phased genotypes, so we know which alleles occur on which of the chromosome pairs. The table lists all the versions of the protein that occur along with their frequencies, including the reference sequence and sequences with one or more alternative alleles.
Click on one of the haplotypes, we’ll go for 18K>*,19del{130}, to find out more about it. Here you will see the frequency in the 1000 Genomes subpopulations, the sequence and the 1000 Genomes individuals where this protein is found.
Let’s have a look at variants in the Location tab. Click on the Location tab in the top bar.
Configure this page and open Variation from the left-hand menu.
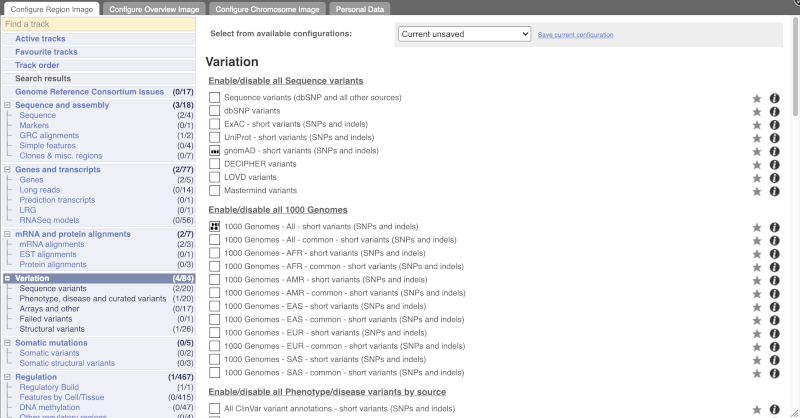
There are various options for turning on variants. You can turn on variants by source, by frequency, presence of a phenotype or by individual genome they were isolated from. You can also turn on genotyping chips.
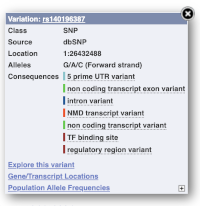
Let’s have a look at a specific variant. If we zoomed in we could see the variant rs334 in this region, however it’s easier to find if we put rs334 into the search box. Click through to open the Variation tab.
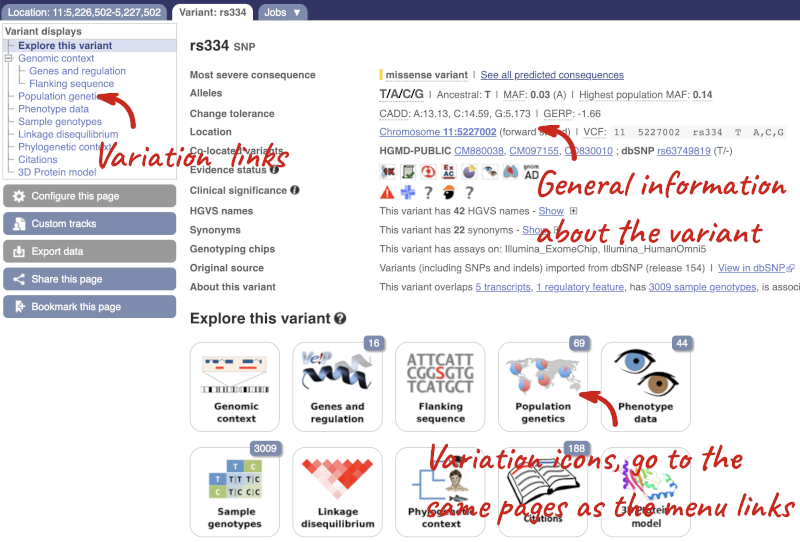
The icons show you what information is available for this variant. Click on Genes and regulation, or follow the link on the left.
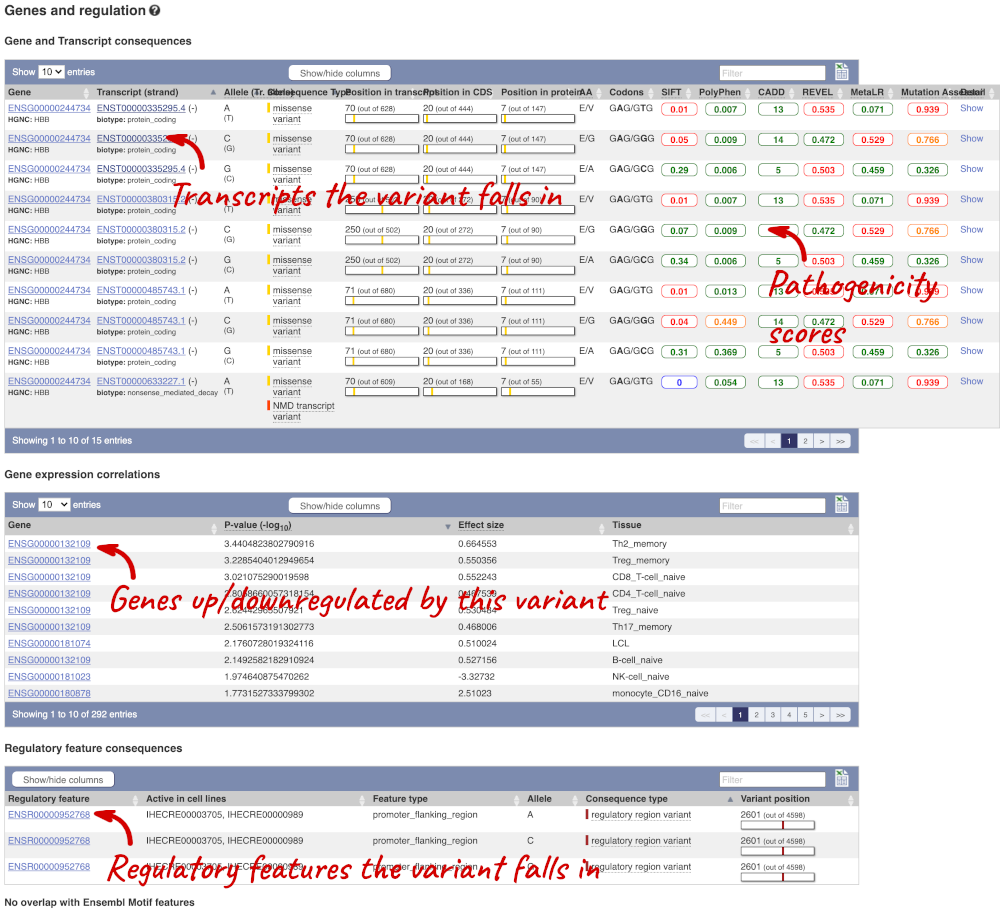
This page illustrates the genes the variant falls within and the consequences on those genes, including pathogenicity predictors. It also shows data from GTEx on genes that have increased/decreased expression in individuals with this variant, in different tissues. Finally, regulatory features and motifs that the variant falls within are shown.
We can also see the variant in the protein structure by clicking on 3D Protein model.
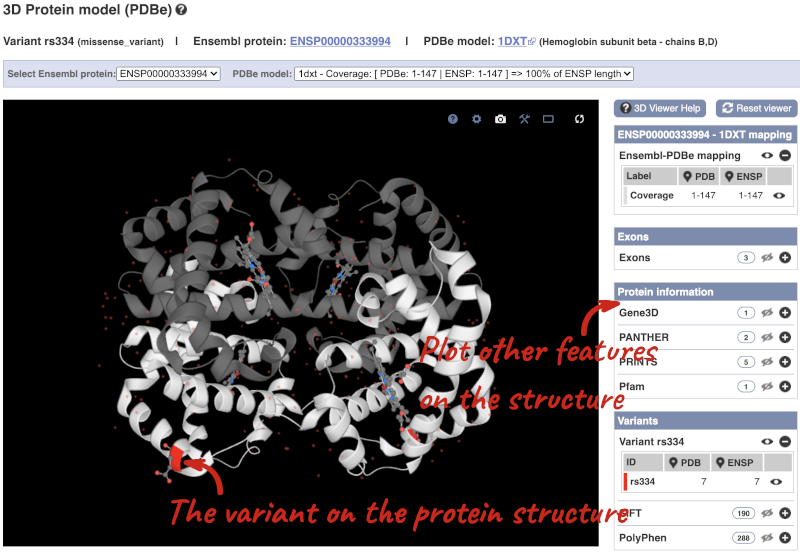
This is a LiteMol viewer, where you can rotate and zoom in on the structure. The variant location is highlighted, so you can see where it lands within the structure.
Let’s look at population genetics. Click on Population genetics in the left-hand menu.
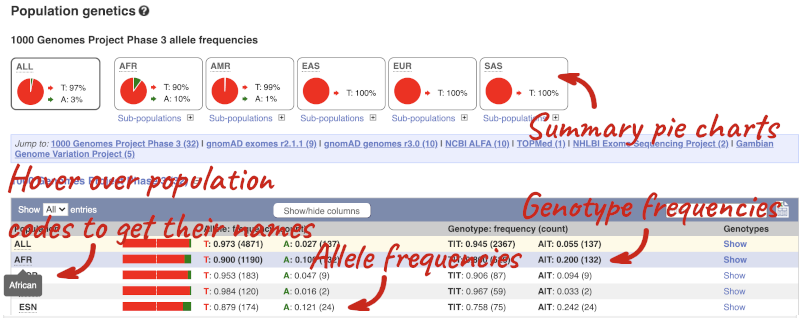
The population allele frequencies are shown by study, including 1000 Genomes and gnomAD. Where genotype frequencies are available, these are shown in the tables.
There are big differences in allele frequencies between populations. Let’s have a look at the phenotypes associated with this variant to see if they are known to be specific to certain human populations. Click on Phenotype Data in the left-hand menu.
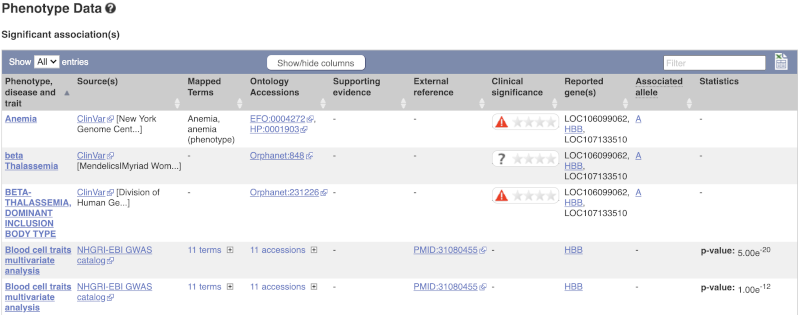
This variant is associated with various phenotypes, including sickle cell and malaria resistance. These phenotype associations come from sources including the GWAS catalog, ClinVar, Orphanet and OMIM. Where available, there are links to the original paper that made the association, the allele that is associated with the phenotype and p-values and other statistics.
Human population genetics and phenotype data
The SNP rs1738074 in the 5’ UTR of the human TAGAP gene has been identified as a genetic risk factor for a few diseases. Use Ensembl to answer the following questions:
-
In which transcripts is this SNP found?
-
What is the least frequent genotype for this SNP in the Yoruba (YRI) population from the 1000 Genomes phase 3?
-
What is the ancestral allele? Is it conserved in the 91 eutherian mammals EPO-Extended?
-
With which diseases is this SNP associated? Are there any known risk (or associated) alleles?
- Please note there is more than one way to get this answer. Either go to the Variation table of the human TAGAP gene, and use the Consequence filter to only include 5’UTR variants, or search Ensembl for
rs1738074directly. Once you’re in the Variant tab, click on Genes and regulation in the menu.This SNP is found in four transcripts of TAGAP. It is also an intron_variant to one lncRNA transcript of TAGAP-AS1.
- Click on Population genetics in the left-hand panel, or click on Explore this variant in the left-hand panel and click the Population genetics icon.
In Yoruba (YRI), the least frequent genotype is CC at the frequency of 5.6%.
- Click on Phylogenetic context in the left-hand panel.
The ancestral allele is T and it’s inferred from the alignment in primates.
Click on Select an alignment which will open a pop-up menu. Open Multiple alignments and select 91 eutherian mammals EPO-Extended. Click on Apply at the bottom of the menu to save your settings.
A region containing the SNP (highlighted in red and placed in the centre) and its flanking sequence are displayed. The T allele is conserved in all but two of the eutherian mammals displayed.
- Click Phenotype data in the left-hand panel.
This variation is associated with multiple sclerosis, celiac disease and white blood cell count. There are known risk alleles for all three diseases and the corresponding P values are provided. The allele A is associated with celiac disease. Note that the alleles reported by Ensembl are T/C. Ensembl reports alleles on the forward strand. This suggests that A was reported on the reverse strand in the original paper. Similarly, one of the alleles reported for Multiple sclerosis is G.
Exploring a SNP in the human genome
The missense variation rs1801133 in the human MTHFR gene has been linked to elevated levels of homocysteine, an amino acid whose plasma concentration seems to be associated with the risk of cardiovascular diseases, neural tube defects, and loss of cognitive function.
-
Find the page with information for rs1801133.
-
Is rs1801133 a missense variant in all transcripts of the MTHFR gene? What is the amino acid change?
-
Why are the alleles for this variation in Ensembl given as G/A and not as C/T, as in the literature?
-
What is the major allele of rs1801133 in different populations?
-
In which paper(s) is the association between rs1801133 and homocysteine levels described?
-
According to the data imported from dbSNP, the ancestral allele for rs1801133 is G. Ancestral alleles in dbSNP are based on a comparison between human and chimp. Does the sequence at this same position in other primates confirm that the ancestral allele is G?
-
Go to the Ensembl homepage. Type
rs1801133in the search box, then click Go. Click on rs1801133. -
Click on Genes and Regulation in the left-hand panel, or click on the Genes and Regulation icon at the top of the page.
No, rs1801133 is missense variant in eight MTHFR transcripts. Please note that this variant is multialleleic with two alternative alleles - as this table displays one consequence per row, each transcript is listed twice.
The amino acid change is A/V for allele A, and A/G for allele C.
-
In Ensembl, the alleles of rs1801133 are given as G/A/C because these are the alleles in the forward strand of the genome. In the literature, the alleles are given as C/T/G because the MTHFR gene is located on the reverse strand. The alleles in the actual gene and transcript sequences are C/T/G. In Ensembl, the allele that is present in the reference genome assembly is always put first.
- Click on Population genetics in the side menu.
In all populations but one, the allele G is the major one. The exception is CLM (Colombian in Medellin; 1000 Genomes).
- Click on Phenotype Data in the left hand side menu.
The specific studies where the association was originally described is given in the Phenotype Data table. Links between rs1801133 and homocysteine levels were described in four papers. Click on the pubmed IDs PMID:34707639, PMID:23696881, PMID:20031578 and PMID:23824729 for more details.
- Click on Phylogenetic Context in the side menu. Select Alignment: 10 primates EPO and click Apply.
Gorilla, bonobo, Sumatran orangutan, chimp, macaque, gibbon, vervet, crab-eating macaque and mouse lemur all have a G in this position.
Exploring a SNP in mouse
In the paper “Altered metabolic signature in pre-diabetic NOD mice” (PloS One. 2012; 7(4): e35445), Madsen et al. have described several regulatory and coding SNPs, some of them in genes involved in ATP and adenosine metabolism, leading to potentially faulty metabolism of ATP and adenosine. The authors describe that one of the identified SNPs in the murine Entpd2 gene (rs28232063) would lead to increased amounts of available ATP, an immune activator, causing increased cell activation and possibly autoreactive T-cell activation. Use Ensembl to answer the following questions:
-
Where is the SNP located (chromosome and coordinates)?
-
What is the HGVS recommendation nomenclature for this SNP?
-
Why does Ensembl put the G allele first (G/A)?
-
Are there differences between the genotypes reported in C57BL/6NJ and NOD/ShiLtJ, according to the Mouse Genomes Project?
- From the Ensembl homepage, select Mouse from the Species search drop-down and enter
rs28232063in the search box.SNP rs28232063 is located on 2:25288362. In Ensembl, its alleles are provided relative to the forward strand.
- Click on Show under HGVS names to reveal information about HGVS nomenclature.
This SNP has got four HGVS names, one at the genomic DNA level (NC_000068.8:g.25288362G>A), two at the transcript level (ENSMUST00000148859.2:n.444-182G>A and ENSMUST00000028328.3:c.446G>A) and one at the protein level (ENSMUSP00000028328.3:p.Arg149Gln).
- In Ensembl, the allele that is present in the reference genome assembly is always put first.
G is the allele for the reference mouse genome strain C57BL/6J
- Click on Sample genotypes is the left-hand panel. The table shows genotypes reported for different mouse strains from the Mouse Genomes Project.
There are indeed differences between the genotypes reported in those two different strains. The genotype reported in C57BL/6NJ is G/G whereas in NOD/ShiLtJ the genotype is A/A.
Exploring VNTR in human
Variable number tandem repeats (VNTRs) show high variation in the number of repeats in the population and are commonly used in forensics (DNA fingerprinting) and to study genetic diversity. (a) Go to the region from 3074666 to 3075100 bp on human chromosome 4. Which gene does it overlap? Which exon of this gene falls in this region?
(b) Configure this page to turn on Repeats (low), Simple repeats (Repeats (low)) and Tandem repeats (TRF) tracks in this view. Can you see any repeats in this exon? What tools were used to annotate the repeats according to the track information?
(c) Zoom in on the (CAG)n to see its sequence. How many CAG repeats can you see in the human reference assembly? Does this track overlap any phenotype-associated variants? What is the identifier of this variant?
(d) Go to the variant tab of the phenotype-associated variant. What is the consequence ontology of this variant? Does the reference allele match the number of repeats you have just counted? What is the shortest and longest allele?
(a) Select Search: Human and type 4:3074666-3075100 in the text box (or alternatively type human 4:3074666-3075100 in the text box). Click Go.
Click on the golden transcript falling in this region. You can see it’s exon 1 of 67 of the huntingtin gene (HTT).
(b) Click Configure this page in the side menu then select: Repeats (low), Simple repeats (Repeats (low)) and Tandem repeats (TRF).
There are three tandem repeats in this exon, and two simple repeats (low); (CAG)n and (CCG)n. Click on the track names to find more about the tools used for annotation: RepeatMasker and Tandem Repeats Finder.
(c) Draw with your mouse a box around the (CAG)n repeat. Click on Jump to region in the pop-up menu.
There are 19 CAG repeats in the human reference sequence overlapping rs71180116 indicated by a pink bar in the All phenotype-associated - short variants (SNPs and indels) track.
(d) Click on the rs71180116 ID to go to the variant tab. You can see in the summary page that this variant is classified as an inframe insertion. Either click + to show all of the alleles in the summary page or go to the Genes and regulation table. This variant has many alternative alleles which differ in the number of repeats. The first allele in the expanded Alleles section of the summary page or the first allele in the Codons column in the Genes and regulation table is the reference allele. It is composed of 19 CAG repeats just as in the Region in detail view. The shortest allele has 7 repeats, the longest has 55 repeats.
VEP
We have identified five variants on human chromosome nine, C-> A at 128203516, an A deletion at 128328461, C->A at 128322349, C->G at 128323079 and G->A at 128322917.
We will use the Ensembl VEP to determine:
- Have my variants already been annotated in Ensembl?
- What genes are affected by my variants?
- Do any of my variants affect gene regulation?
Go to the front page of Ensembl and click on the Variant Effect Predictor.

This page contains information about the VEP, including links to download the script version of the tool. Click on Launch VEP to open the input form:
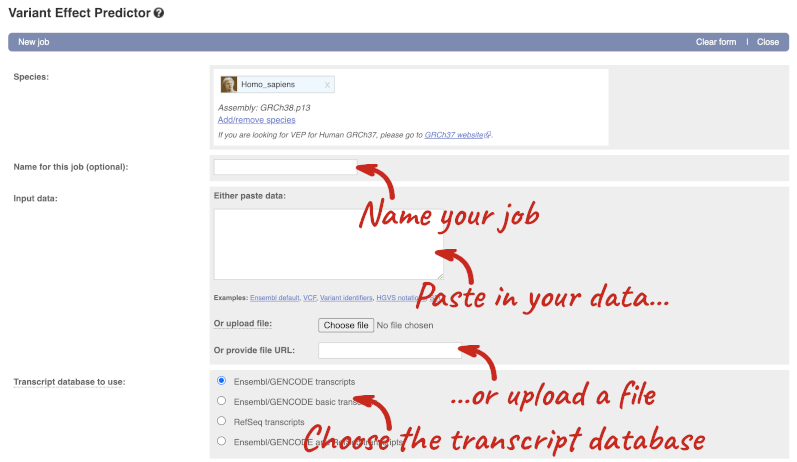
The data is in VCF format:
chromosome coordinate id reference alternative
Put the following into the Paste data box:
9 128328460 var1 TA T
9 128322349 var2 C A
9 128323079 var3 C G
9 128322917 var4 G A
9 128203516 var5 C A
The VEP will automatically detect that the data is in VCF.
There are further options that you can choose for your output. These are categorised as Identifiers, Variants and frequency data, Additional annotations, Predictions, Filtering options and Advanced options. Let’s open all the menus and take a look.
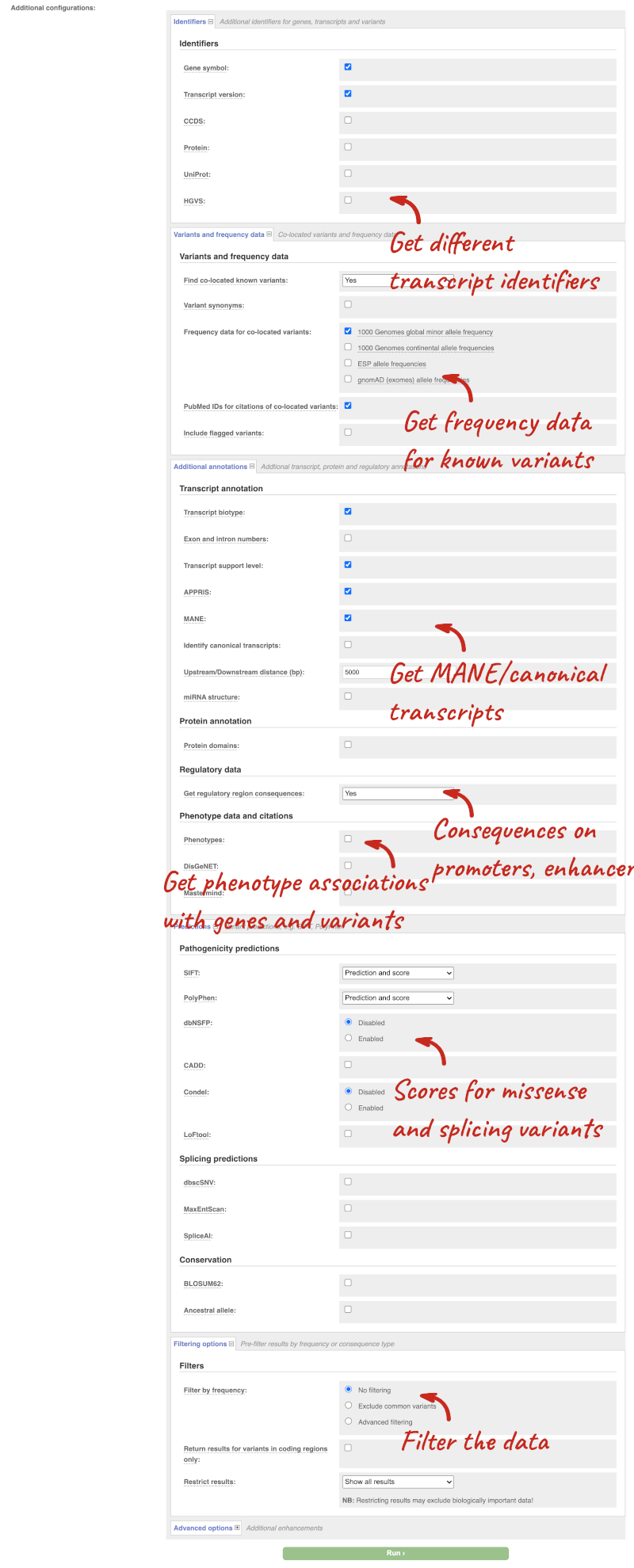
Hover over the options to see definitions.
We’re going to select some options:
- HGVS, annotation of variants in terms of the transcripts and proteins they affect, commonly-used by the clinical community
- Phenotypes
- Protein domains
When you’ve selected everything you need, scroll right to the bottom and click Run.

The display will show you the status of your job. It will say Queued, then automatically switch to Done when the job is done, you do not need to refresh the page. You can edit or discard your job at this time. If you have submitted multiple jobs, they will all appear here.
Click View results once your job is done.
In your results you will see a graphical summary of your data, as well as a table of your results.
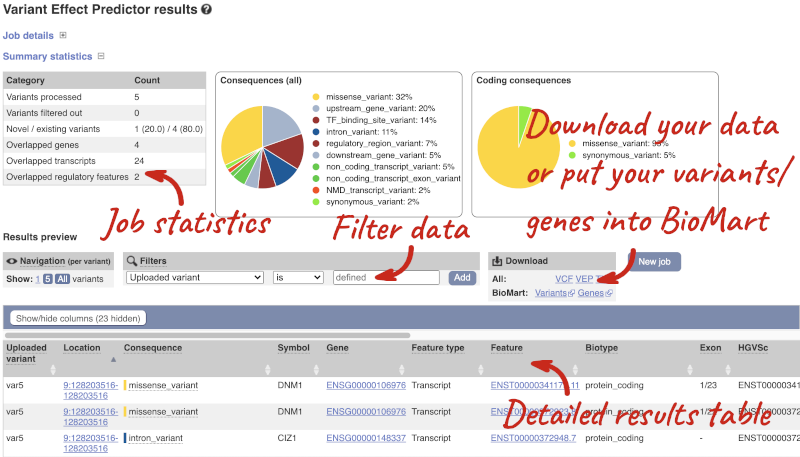
The results table is enormous and detailed, so we’re going to go through the it by section. The first column is Uploaded variant. If your input data contains IDs, like ours does, the ID is listed here. If your input data is only loci, this column will contain the locus and alleles of the variant. You’ll notice that the variants are not neccessarily in the order they were in in your input. You’ll also see that there are multiple lines in the table for each variant, with each line representing one transcript or other feature the variant affects.
You can mouse over any column name to get a definition of what is shown.
The next few columns give the information about the feature the variant affects, including the consequence. Where the feature is a transcript, you will see the gene symbol and stable ID and the transcript stable ID and biotype. Where the feature is a regulatory feature, you will get the stable ID and type. For a transcription factor binding motif (labelled as a MotifFeature) you will see just the ID. Most of the IDs are links to take you to the gene, transcript or regulatory feature homepage.
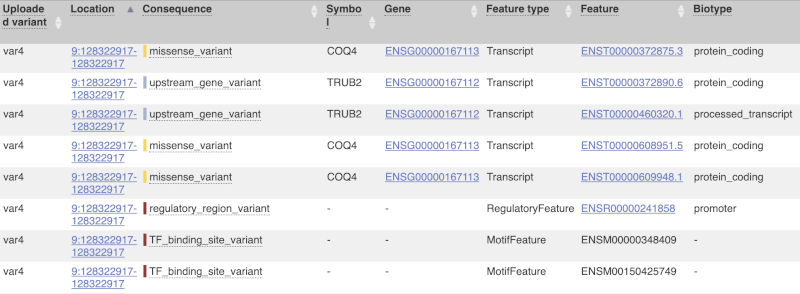
This is followed by details on the effects on transcripts, including the position of the variant in terms of the exon number, cDNA, CDS and protein, the amino acid and codon change, transcript flags, such as MANE, on the transcript, which can be used to choose a single transcript for variant reporting, and predicted pathogenicity scores. The predicted pathogenicity scores are shown as numbers with coloured highlights to indicate the prediction, and you can mouse-over the scores to get the prediction in words. Two options that we selected in the input form are the HGVS notation, which is shown to the left of the image below and can be used for reporting, and the Domains to the right. The Domains list the proteins domains found, and where there is available, provide a link to the 3D protein model which will launch a LiteMol viewer, highlighting the variant position.
![]()
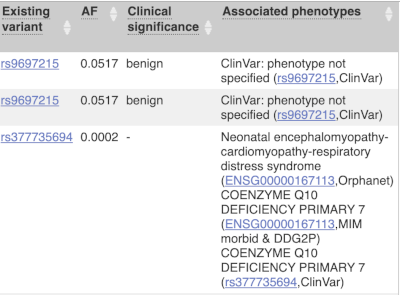
Where the variant is known, the ID of the existing variant is listed, with a link out to the variant homepage. In this example, only rsIDs from dbSNP are shown, but sometimes you will see IDs from other sources such as COSMIC. The VEP also looks up the variant from frequency files, and pulls back the allele frequency (AF in the table), which will give you the 1000 Genomes Global Allele Frequency. In our query, we have not selected allele frequencies from the continental 1000 Genomes populations or from gnomAD, but these could also be shown here. We can also see ClinVar clinical significance and the phenotypes associated with known variants or with the genes affected by the variants, with the variant ID listed for variant associations and the gene ID listed for gene associations, along with the source of the association.
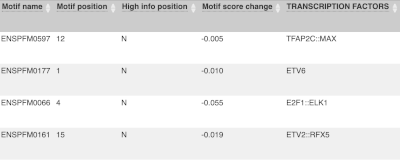
For variants that affect transcription factor binding motifs, there are columns that show the effect on motifs (you may need to click on Show/hide columns at the top left of the table to display these). Here you can see the position of the variant in the motif, if the change increases or decreases the binding affinity of the motif and the transcription factors that bind the motif.
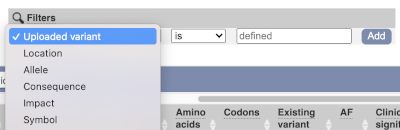
Above the table is the Filter option, which allows you to filter by any column in the table. You can select a column from the drop-down, then a logic option from the next drop-down, then type in your filter to the following box. We’ll try a filter of Consequence, followed by is then missense_variant, which will give us only variants that change the amino acid sequence of the protein. You’ll notice that as you type missense_variant, the VEP will make suggestions for an autocomplete.
You can export your VEP results in various formats, including VCF. When you export as VCF, you’ll get all the VEP annotation listed under CSQ in the INFO column. After filtering your data, you’ll see that you have the option to export only the filtered data. You can also drop all the genes you’ve found into the Gene BioMart, or all the known variants into the Variation BioMart to export further information about them.
Running CFTR variants through VEP
Resequencing of the genomic region of the human CFTR (cystic fibrosis transmembrane conductance regulator (ATP-binding cassette sub-family C, member 7) gene (ENSG00000001626) has revealed the following variants. The alleles defined in the forward strand:
- G/A at 7: 117,530,985
- T/C at 7: 117,531,038
- T/C at 7: 117,531,068
Use the VEP tool in Ensembl and choose the options to see SIFT and PolyPhen predictions. Do these variants result in a change in the proteins encoded by any of the Ensembl genes? Which gene? Have the variants already been found?
Go to the Ensembl homepage and click on the link Tools at the top of the page. Currently there are nine tools listed in that page. Click on Variant Effect Predictor and enter the three variants as below:
7 117530985 117530985 G/A
7 117531038 117531038 T/C
7 117531068 117531068 T/C
Note: Variation data input can be done in a variety of formats. See more details about the different data formats and their structure in this VEP documentation page. Click Run. When your job is listed as Done, click View Results.
You will get a table with the consequence terms from the Sequence Ontology project (http://www.sequenceontology.org/) (i.e. synonymous, missense, downstream, intronic, 5’ UTR, 3’ UTR, etc) provided by VEP for the listed SNPs. You can also upload the VEP results as a track and view them on Location pages in Ensembl. SIFT and PolyPhen are available for missense SNPs only. For two of the entered positions, the variations have been predicted to have missense consequences of various pathogenicity (coordinate 117531038 and 117531068), both affecting CFTR. All the three variants have been already annotated and are known as rs1800077, rs1800078 and rs35516286 in dbSNP (databases, literature, etc).
VEP analysis of structural variants in human
We have details of a genomic deletion in a breast cancer sample in VCF format:
13 32307062 sv1 . <DEL> . . SVTYPE=DEL;END=32908738
Use VEP in Ensembl to find out the following information:
1. How many genes have been affected?
2. Does the structural variant (SV) cause deletion of any complete transcripts?
3. Map your variant in the Ensembl browser on the Region in detail view.
- Click on VEP at the top of any Ensembl page and open the web interface. Make sure your species is Human. It is good practise to name your VEP jobs something descriptive, such as Patient deletion exercise. Paste the variant in VCF format into the Paste data field and hit Run.
12 different genes are affected by the SV.
- Filter your table by select Consequence is
transcript_ablationat the top of the table and click Add.Yes, there is deletion of complete transcripts of PDS5B, N4BP2L1, BRCA2, RNY1P4, IFIT1P1, ATP8A2P2, N4BP2L2, N4BP2L2-IT2 and one gene without official symbols: ENSG00000212293.
- To view your variant in the browser click on the location link in the results table 13: 32307062-32908738. The link will open the Region in detail view in a new tab. If you have given your data a name it will appear automatically in red. If not, you may need to Configure this page and add it under the Personal data tab in the pop-up menu.
Comparative genomics
Let’s look at the homologues of human BRCA2. Search for the gene and go to the Gene tab.
Click on Gene tree, which will display the current gene in the context of a phylogenetic tree used to determine orthologues and paralogues.
.png)
Funnels indicate collapsed nodes. We can expand them by clicking on the node and selecting Expand this sub-tree from the pop-up menu.
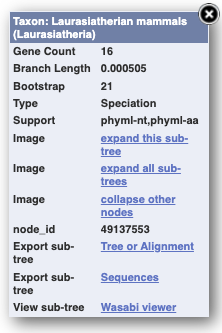
We can also see the alignment of the sub-tree by clicking on Wasabi viewer, which will open a pop-up:

You can download the tree in a variety of formats. Click on the download icon in the bar at the top of the image to get a pop-up where you can choose your format.

We can look at homologues in the Orthologues and Paralogues pages, which can be accessed from the left-hand menu. If there are no orthologues or paralogues, then the name will be greyed out. Paralogues is greyed out for BRCA2 indicating that there are no paralogues available. Click on Orthologues to see the 175 orthologues available.
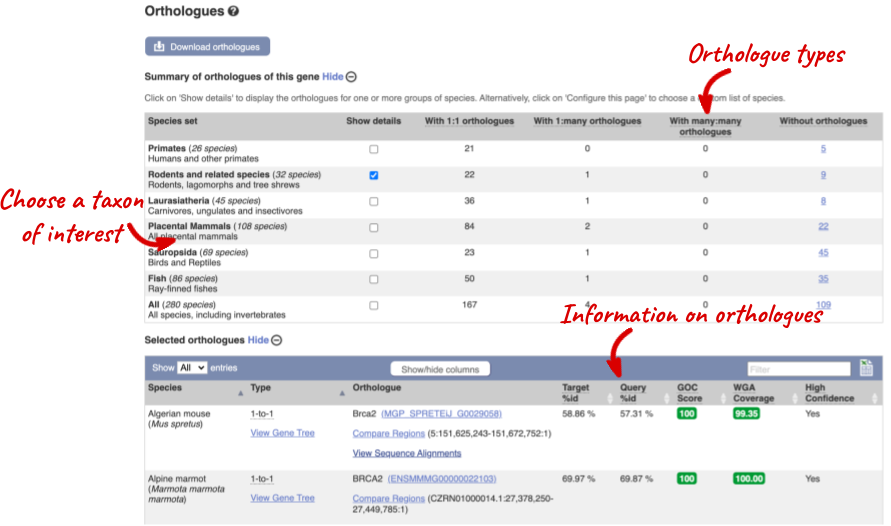
Choose to see only Rodents and related species orthologues by selecting the box. The table below will now only show details of rodent orthologues. Let’s look at mouse.

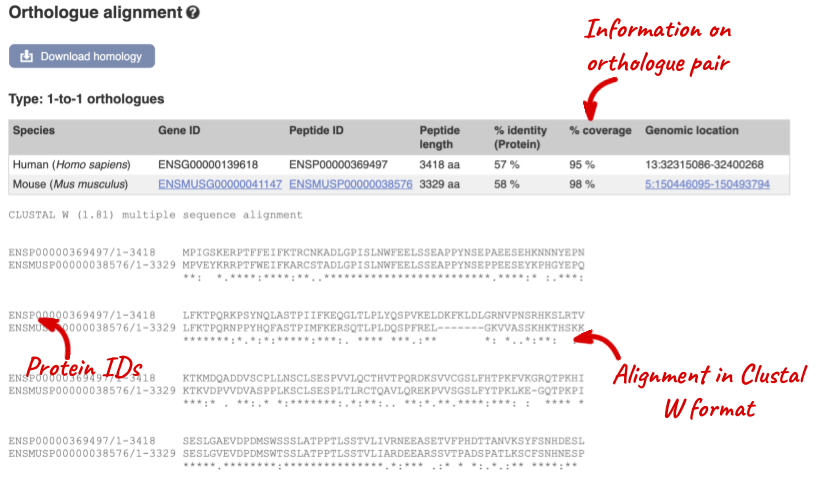
Links from the orthologue allow you to go to alignments of the orthologous proteins and cDNAs. Click on View Sequence Alignments then View Protein Alignment for the mouse orthologue.
Let’s look at some of the comparative genomics views in the Location tab. Go to the region 2:176087000-176202000 in human, which contains the HoxD cluster which is involved in limb development and is highly conserved between species.
You can turn on conservation scores and constrained elements. Click on Configure this page, then Comparative genomics and turn on the tracks for Constrained elements for 91 eutherian mammals EPO-Extended and Conservation score for 91 eutherian mammals EPO-Extended. Save and close the menu.
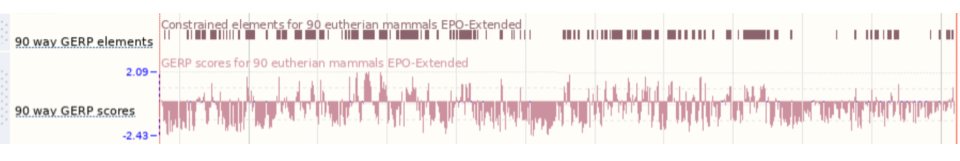
You can now see the conservation scores in pale pink. These were used to determine the peaks indicated in the constrained elements track in dark pink. This track indicates regions of high conservation between species, considered to be “constrained” by evolution.
We can also look at individual species comparative genomics tracks in this view by clicking on Configure this page.
Select BLASTz/LASTz alignments from the left-hand menu to choose alignments between closely related species. Turn on the alignments for Mouse, Chicken and Chimpanzee in Normal. Save and close the menu.
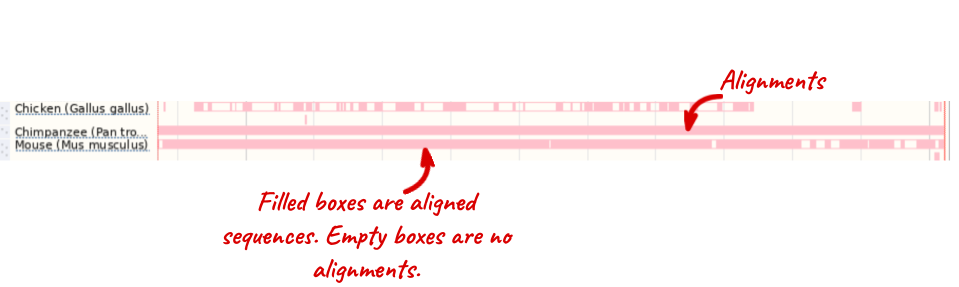
The alignment is greatest between closely related species.
We can also look at the alignment between species or groups of species as text. Click on Alignments (text) in the left hand menu.
Select Select an alignment to open the alignment menu.
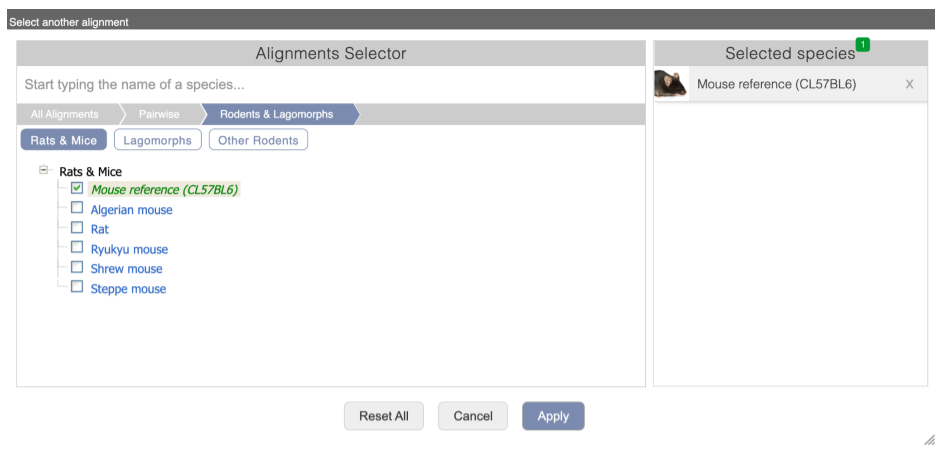
Click through the links, Pairwise, Rodents & Lagomorphs, Rats and Mice to select Mouse reference (CL57BL6).
In this case there are two blocks aligned, Block 1 a large (115001 bp) alignment against mouse chr2 and one smaller block against mouse chr7. Click on Block 1.
You will see a list of the regions aligned, followed by the sequence alignment. Click on Display full alignment. Exons are shown in red.
To compare with both contigs visually, go to Region comparison.
To add species to this view, click on the blue Select species or regions button. Choose Mouse Reference again then close the menu.
.png)
You can configure this view for both species. Click on Configure this page and look in the top left of the menu.

The drop down allows you to configure each species separately.
We can view large scale syntenic regions from our chromosome of interest. Click on Synteny in the left hand menu.
.png)
Orthologues and gene trees for the human BRAF gene
Go to Ensembl to answer the following questions:
-
How many orthologues are predicted for the human BRAF in primates? How much sequence identity does the Carlito syrichta (tarsier) protein have to the human one? Can you tell which end of the BRAF protein is more conserved between these two species by looking at the orthologue alignment?
-
Go to the Gene tree for this gene. View the Wasabi alignment of all the proteins in primates. Can you see a large gap in the alignment around position 450? Which species match the human sequence?
- From the Ensembl homepage, choose Human from the drop-down list and search for
BRAF. Click through to the Gene tab view. Click on Orthologues at the left side of the page to see all the orthologous genes.There are 1:1 orthologues in 22 primates reported in the summary table.
Search for
Tarsierin the table below.The percentage of identical amino acids in the tarsier protein (the orthologue) compared with the gene of interest. i.e. human BRAF (the target species/gene) is 95.39%. This is known as the Target%id. The identity of the gene of interest (human BRAF) when compared with the orthologue (tarsier BRAF, the query species/gene) is 94.65% (the Query %id). Note the difference in the values of the Target and Query %id reflects the different protein lengths for the human and tarsier BRAF genes.
Click on the View Sequence Alignments link in the Orthologue column to View Protein Alignment in Clustal W format.
Conserved amino acids are indicated by asteriks. The alignment around the N-terminus looks poorer, when compared to the C-terminus end.
- Click on Gene tree in the left hand menu. All of the primates are enclosed in a lilac box. Click on the furthest left node in the box to get a pop-up labelled Primates. Alternatively, scroll to the bottom of the page, and select Order from Collapse all the nodes at the taxonomic rank. Primates will appear as a red triangle. Click on Wasabi viewer in the pop-up menu to see the alignment. Scroll to position 450.
Greater bamboo lemur, mouse lemur, Sumatran orangutan, crab-eating macaque, olive baboon, Bolivian squirrel monkey, white-tufted-ear marmoset and Ma’s night monkey all match the human sequence.
Cow orthologues
Find the ABCC11 gene on the cow genome. (a) Go to the Location tab for this gene. View the Alignments (image) for the 43 eutherian mammals EPO. Do all the mammals have an alignment in this region? Can you spot a difference in the alignment between Pecora (including cattle, goats, sheep and deers) and the remaining mammals?
(b) Let’s now see this alignment as text. Go to Alignments (text) for the 43 eutherian mammals EPO. Sort the aligned blocks by genomic coordinates and view the 3’ portion of the ABCC11 gene (smallest coordinates). Does it support your previous conclusions? Export the alignment without ancestral sequences as ClustalW.
(c) Click on the Region in detail link at the left and turn on the tracks for 90 eutherian mammals EPO-Extended, Constrained elements and Conservation score for the 90 eutherian mammals EPO-Extended by configuring the page. What is the difference between the Multiple alignment track and the Constrained elements track? Which regions of the gene do most of the constrained element blocks match up to? Can you find more information on how the Constrained elements track was generated?
(a) Search for cow ABCC11 from the home page. Click on ABCC11 genomic coordinates 18:16590653-16667410:-1 in the search results to open the Location tab. Click on Alignments (image) at the left, and select the 43 eutherian mammals EPO multiple alignment by clicking on Select an alignment blue button. Scroll down to see the hidden and missing species.
All but 13 of the 43 mammals have an alignment at this region. ABCC11 gene model for the closely related Pecora species (cows, yak, goat, sheep and Yarkand deer) is longer when compared to the other species, with many additional exons at its 3’ end (left side of the image), which are absent in other taxa.
(b) Click on Alignments (text) in the left hand menu. The 43 eutherian mammals EPO alignment should be pre-selected. Scroll down to the table of alignment blocks. Sort the table by clicking on small arrows in the Location on Cow column header. The alignment blocks are now sorted by the genomic coordinates, with smalles coordiantes corresponding to the 3’ most end of ABCC11 (located on the reverse strand). Click on Block 3 to view the alignment.
Only 5 species have an alignment in this region, including cows, yak, goat, sheep and Yarkand deer, which is in agreement with our previous observation. Scroll up and click on Download alignment blue button, change File format to CLUSTALW, then Download.
(c) Click on Region in detail in the left hand menu. Turn on the 90 eutherian mammals EPO-Extended, Constrained elements and Conservation score for 90 eutherian mammals EPO-Extended tracks, all under the Comparative genomics in the Configure this page menu.
The 90 eutherian mammals EPO-Extended multiple alignment track is shown as pink block indicating that the whole region can be aligned at this locus. The GERP elements and GERP scores tracks show where the conserved sequence is located in the alignment. Conserved elements shown as pink boxes match up with exonic regions of the 5’-half of this cow gene (right side of the image). In general, exons tend to be highly conserved across taxa. Click on the track name and the i icon (information button) to read more about constrained elements (or any other data track).
Synteny
Start at Ensembl homepage.
-
Find the rhodopsin (RHO) gene in human. Go to the Location tab and click Synteny at the left. Are there any syntenic regions in duck? If so, which chromosomes are shown in this view?
-
Stay in the Synteny view. Is there a homologue in duck for human RHO? Are there more genes in this syntenic block with homologues? Which duck chromosome is this human genomic region syntenic to?
- Search for
human RHOfrom the home page. Click on RHO genomic coordinates 3:129528639-129535344:1 in the search results to directly open the Location tab. Click Synteny at the left and change the species to Duck next to the image.Yes, there are multiple syntenic regions in duck to human chromosome 3, which is in the centre of this view. Duck chromosomes 1, 2, 7, 9, 13, and Z have syntenic regions to human chromosome 3.
- Scroll down to the bottom of the page to see a list of homologous genes on both genomes.
Human RHO is homologous to RHO (ENSAPLG00000005189) in duck. Click 15 upstream genes or 15 downstream genes to view neighbouring genes in this syntenic block. There are many neighbouring genes with homologues in duck. Human genes in this region are homologous to duck genes on chromosome 13, which is also indicated by the red boxes in the image above as this genomic block on human chromosome 3 is syntenic to duck chromosome 13.
Whole genome alignments
(a) Find the human BRCA2 gene and go to the Region in detail page. Turn on the BLASTz/LASTz alignment tracks for chicken, chimp, mouse and platypus. Does the degree of conservation between human and the various other species reflect their evolutionary relationship? Which parts of the BRCA2 gene seem to be the most conserved? Did you expect this?
(b) Have a look at the Conservation score and Constrained elements tracks for the set of 90 eutherian mammals and 65 amniota vertebrates. Do these tracks confirm what you already saw in the pairwise alignment tracks?
(c) Retrieve the genomic alignment (text) across 65 amniotes for a constrained element matching up with exon 15 of the golden transcript. Highlight the bases that match in >50% of the species in the alignment. Is this sequence exonic in all species?
(a) Select Human from the species selector drop-down list and type brca2 in the search box. Click Go. Click on 13:32315086-32400268:1 below BRCA2 (Human Gene) to go to the Region in detail page.
Click Configure this page in the side menu, then BLASTz/LASTz alignments under the Comparative genomics menu. Select Chicken, Chimpanzee, Mouse and Platypus in Normal style.
Yes, the degree of conservation does reflect the evolutionary relationship between human and the other species; the highest degree of conservation is found in chimp, followed by mouse, platypus and chicken, respectively.
Especially the exonic sequences of BRCA2 seem to be highly conserved between the various species, which is what is to be expected because these are supposed to be under higher selection pressure than intronic and intergenic sequences.
(b) Click Configure this page in the side menu, then Conservation regions under the Comparative genomics menu.
Select Conservation score and Constrained elements for 90 eutherian mammals EPO-Extended and 65 amniota vertebrates Mercator-Pecan.
Both the Conservation score and Constrained elements tracks largely correspond with the data seen in the pairwise alignment tracks; all exons of the BRCA2 gene show a high degree of conservation (note the UTRs which are not conserved).
(c) Click on exons of the golden transcript (ENST00000380152) to reveal their rank in transcript. Exon 15 can be found in the middle. Click on a constrained element in 65 way GERP elements track matching up with this exon.
Click on View alignments (text) in the pop-up menu, then Configure this page in the side menu. Select Show conservation regions to highlight bases matching in majority of the species in this alignment.
Exons are indicated by red lettering. All but Naja naja (Indian cobra) and Pseudonaja textilis (Eastern brown snake) have exonic sequence in this region.
Regulation
We’re going to look for regulatory features in the region of a gene and investigate their activity in different cell types. We’ll start by searching for the gene KPNA2 and jumping to the Location tab. Scroll down to the Region in detail view and zoom out a little to see the gene as well as its flanking regions.
The Regulatory Build track is shown by default.

In this region we can see a number of regulatory features, including a red promoter with light red promoter flanks, cyan CTCF binding sites, yellow enhancers and lilac transcription factor (TF) binding sites (don’t worry if you have zoomed out further or not as far and can see more/less). Refer to the legend at the bottom of the view to see what each of the colours mean.
You can also click on the individual regulatory features to learn more. Click on the red promoter to open a pop-up menu.

Click on the stable ID, ENSR00000097453, to jump to the Regulation tab.
Here, you can find a summary of the activity of the promoter in the different cell types. Scroll down to Summary of Regulatory Aactivity to find out in which cells the promoter is active (the feature displays an active epigenetic signature, which can include evidence of open chromatin), inactive (the region bears no epigenetic modifications from the ones included in the Regulatory Build), poised (the feature displays a epigenetic signature with the potential to be activated) or repressed (the feature is epigenetically suppressed). We can see that this promoter is active in one out of the 118 cell types currently in Ensembl.
Let’s switch back to the Location tab to explore the different regulation tracks that are available. Click on Configure this page and in the pop-up window under the Regulation section, click on Other regulatory regions and enable the Fantom 5, TarBase and Motif features tracks. Close the pop-up window.
The Fantom 5 track displays transcription start site (TSS) and enhancer predictions from the FANTOM5 project.
The TarBase track displays experimentally verified miRNA targets from TarBase.
The Motif features track indicates the positions of transcription factor binding motifs (TFBMs) in black lines/blocks. You can click on individual features to find out more information about the TFBM, including a list of TFs binding at this site and, if available, in which cells the TFBM was experimentally verified in. You can also view the Binding matrix** by clicking on the matrix ID. This opens a pop-up window which displays the binding matrix used and a binding score representing how well a particular site matches the binding matrix.
We can explore more detailed data by adding further Regulation tracks. Click on the Configure this page button on the left-hand side.

In the pop-up window, go to Regulation and click on Features by Cell/Tissue to view the detailed activity of the regulatory feature by cell type.
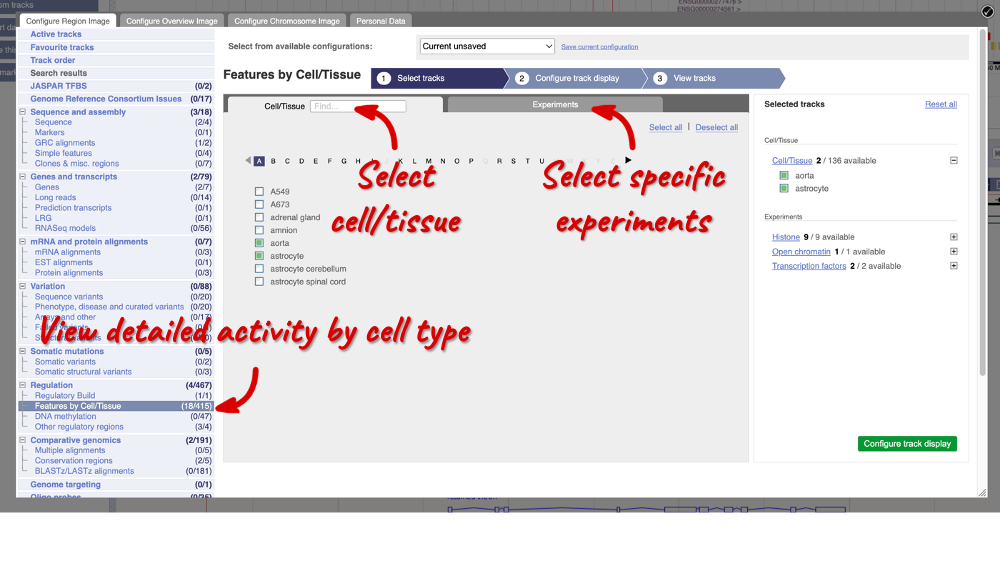
We can add cells by clicking on them. Find them using the search or the alphabet ribbon. Let’s add a cell type where the promoter is inactive, aorta, and one where it’s active astrocytes. Once you’ve selected the cells, they will appear in the menu on the right, where you can easily view the list by clicking on the + icon and de-select them.
To choose the experiments to see data on, click on the Experiments tab at the top of the menu. You can navigate this the same as the Cell/Tissue tab, except that you have to choose between Histone, Open Chromatin and Transcription factors. Let’s Select all in all categories.
When you’ve chosen your experiments and cells, you can click on the green Configure track display button in the bottom right-hand corner.
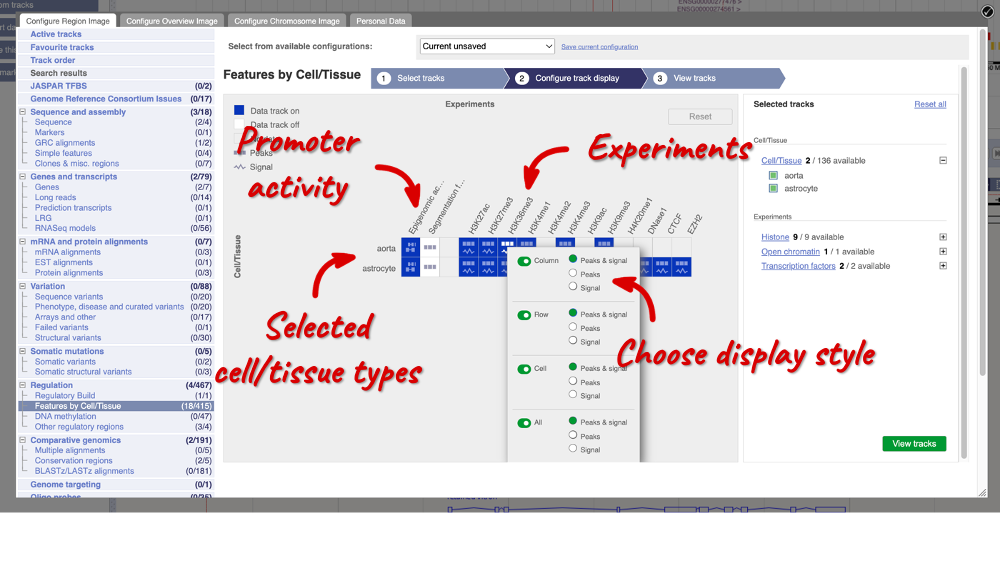
Now we can see the active feature in astrocytes compared to the inactive feature in aorta.
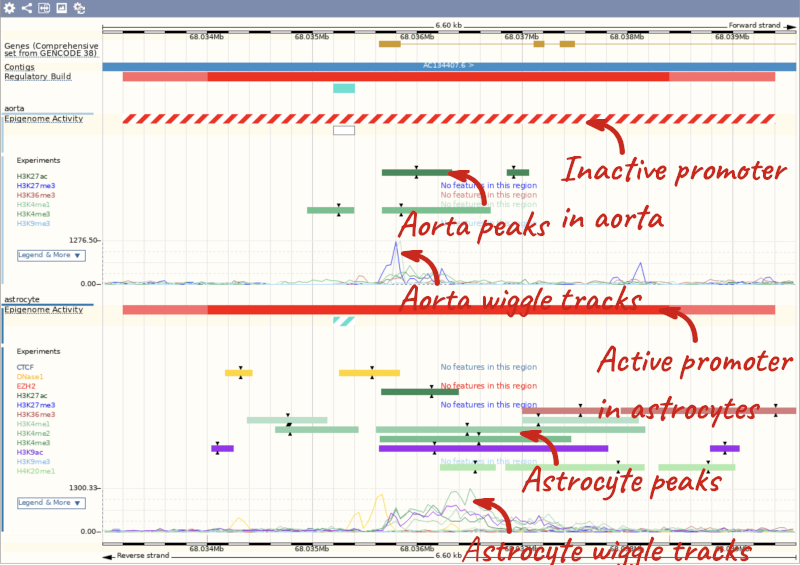
Regulatory features between INSIG1 and BLACE in human
-
Find the Location tab (Region in detail view) for the region between the genes INSIG1 and BLACE. Are there any predicted enhancers in this region?
-
Go to the Regulation tab for the enhancer ENSR00001133586. How many cell types is this enhancer active in? Are there any cell types where its activity is repressed?
-
Switch to the Location tab. Take a look at the histone modifications across this enhancer in neutro myelocyte cells, where this enhancer is active, compared to neutrophil (CB) cells, where it is poised. What differences can you observe?
-
Are there any verified transcription factor binding motifs in this enhancer? In what cells?
- Search for
human INSIG1from the Ensembl homepage. Click on INSIG1 genomic coordinates 7:155297776-155310235:1 in the search results to open the Location tab directly. In the Region overview display, drag out a box to encompass the neighbouring BLACE gene. Scroll down to the Region in detail display. Have a look at the Regulatory Build track. You can find a legend of this track underneath the display.There are 5 yellow enhancers in the region between the genes INSIG1 and BLACE.
- There are several ways to search for the enhancer. You can click the different enhancer features in the Regulatory Build track to find ENSR00001133586, or you can search Ensembl for the ID ENSR00001133586 and navigate to the Regulation tab. Under the Activity display, you can find the activity of the regulatory feature across different cell types.
ENSR00001133586 is active in neutro myelocyte cells only. It is repressed in 34 cell types.
- Click on the Location tab. Choose cells by clicking on the Configure this page button on the left-hand panel or Add/remove tracks button above the Region in detail display. In the pop-up window, click on Features by Cell/Tissue in the left-hand menu. Select neutro myelocyte in which this enhancer is active and neutrophil (CB) in which it is poised. Add experiment tracks by clicking on Experiments tab and Select all under Histone. Click Configure track display, then View tracks to load the page.
Both cell types have H3K27me3, H3K4me1 and H3K9me3 histone modifications at this locus, while neutro myelocyte cells also have H3K27ac and H3K36me3 modifications, and neutrophils (CB) have H3K4me3 modifications. The different clusters of peaks indicate different epigenetic profiles, which might explain the difference in the enhancer activity between these two cell types.
- Stay in the Location tab. Click on the Configure this page button on the left-hand panel or Add/remove tracks button above the Region in detail display. In the pop-up window in the left-hand menu, go to the Regulation section and click on Other regulatory regions. Enable the Motif features track to visualise any transcription factor (TF) binding motifs. Close the pop-up window. Find the Motif features track. There are two black markers indicating verified TF motifs. Click on them to tell which motifs and which cells.
The two motifs are both verified in K562 cells and bind a number of different TFs. The ENSM00523362328 motif binds ELF1, ELF2, ELK1, FLI1, ERG, ETS1, ETV6, FOXO1::ELK3, FOXO1::ETV1, ETV1, ETV2, ERF, ELK3, ETV3, GABPA, ETS2, ELK4, FEV, ETV5 and ETV4. ENSM00523900117 binds ETV7, ETS1 and ELK1::SPDEF.
Regulatory features in human
-
Search for the regulatory feature ENSR00000262400. What type of feature is this? What is its genomic location?
-
Which cell types is this feature inactive and/or repressed in? View the supporting evidence for the repressed cell type. What project was the repressed cell type studied in?
-
Why do so many cells have this feature listed as NA on the Activity display?
-
Search for
ENSR00000262400on the Ensembl homepage. Click on the search result to open the Regulation tab.
ENSR00000262400 is a CTCF binding site found at Chromosome 11: 1,998,001 - 2,001,400, which can be found at the top of the Activity page. -
Scroll down to see the summary of regulatory activity across different cell types.
The CTCF binding site is inactive in H1-hESC_3 and HepG2 cells. It is repressed in A673.Click on Source Data at the top of the page or in the left-hand menu. Use the filter at the top right-hand corner of the table and enter A673. You can find the source of the supporting evidence under the Source column. The cell type A673 was studied in the ENCODE project.
-
Note that many cell types have this feature represented as NA. This is because no corresponding CTCF signal or peaks are available for these cell types as they were not studied in the project sources.
Cells which do not have CTCF ChIP-seq data cannot have an activity listed for this feature.
BioMart
Follow these instructions to guide you through BioMart to answer the following query:
You have three questions about a set of human genes:
ESPN, MYH9, USH1C, CISD2, THRB, WHRN
(these are HGNC gene symbols. More details on the HUGO Gene Nomenclature Committee can be found on http://www.genenames.org)
- What are the NCBI Gene IDs for these genes?
- Are there associated functions from the GO (gene ontology) project that might help describe their function?
- What are their cDNA sequences?
Click on BioMart in the top header of a www.ensembl.org page to go to: www.ensembl.org/biomart/martview
You cannot choose any filters or attributes until you’ve chosen your dataset. Your dataset is the data type you’re working with. In this case we’re going to choose human genes, so pick Ensembl Genes then Human genes from the drop-downs.
Now that you’ve chosen your dataset, the filters and attributes will appear in the column on the left. You can pick these in any order and the options you pick will appear.
Click on Filters on the left to see the available filters appear on the main page. You’ll see that there are loads of categories of Filters to choose from. You can expand these by clicking on them. For our query, we’re going to expand GENE.
Our input data is a list of identifiers, so we’re going to use the Input external references ID list filter. This allows us to input a list of identifiers from different databases. We need to choose what kind of identifier we’re using, so that BioMart can look up the right column in a data table. You can pick these from a drop-down list, which lists the type of identifier with an example of how it looks. For our query, we have a list of gene names, so we need to pick Gene Name(s).
To check if the filters have worked, you can use the Count button at the top left, which will show you how many genes have passed the filter. If you get 0 or another number you don’t expect, this can help you to see if your query was effective.
To choose the attributes, expand this in the menu. There are six categories for human gene attributes. These categories are mutually exclusive, you cannot pick attributes from multiple categories. This means that we need to do two separate queries to get our GO terms and NCBI IDs, and to get our cDNA sequences.

The Ensembl gene and transcript IDs, with and without version numbers are selected by default. The selected attributes are also listed on the left.

We can choose the attributes we want by clicking on them. For our query, we’re going to select:
- GENE
- Gene Name
- EXTERNAL
- NCBI gene ID
- GO term accession
- GO term name
- GO term definition
We need to select the Gene Name in order to get back our original input, as this is not returned by default in BioMart. The order that you select the attributes in will define the order that the columns appear in in your output table.
You can get your results by clicking on Results at the top left.
The results table just gives you a preview of the first ten lines of your query. This allows the results to load quickly, so that if you need to make any changes to your query, you don’t waste any time. To see the full table you can click on View ## rows. You can also export the data to an xls, tsv, csv or html file. For large queries, it is recommended that you export your data as Compressed web file (notify by email), to ensure your download is not disrupted by connection issues.
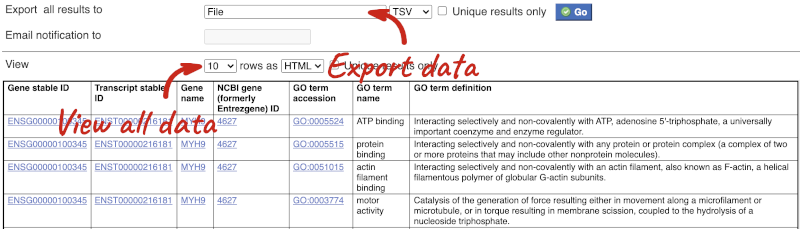
You can see multiple rows per gene in your input list, because there are multiple transcripts per gene and multiple GO terms per transcript.
To get the cDNA sequences, go back to the Attributes then select the category Sequences and expand SEQUENCES.
When you select the sequence type, the part of the transcript model you’ve chosen will be highlighted in the grpahic.
Choose cDNA sequences, then expand HEADER INFORMATION to add Gene Name to the header. Then hit Results again.

For more details on BioMart, have a look at this publication:
Kinsella, R.J. et al
Ensembl BioMarts: a hub for data retrieval across taxonomic space.
http://europepmc.org/articles/PMC3170168
Finding genes by protein domain
Find mouse proteins with Signalp cleavage sites located on chromosome 9.
As with all BioMart queries you must select the dataset, set your filters (input) and define your attributes (desired output). For this exercise: Dataset: Ensembl genes in mouse Filters: Signalp cleavage sites on chromosome 9 Attributes: Ensembl gene and transcript IDs and gene names
Go to the Ensembl homepage (http://www.ensembl.org) and click on BioMart at the top of the page. Select Ensembl genes as your database and Mouse genes as the dataset. Click on Filters on the left of the screen and expand REGION. Change the chromosome to 9. Now expand PROTEIN DOMAINS, also under filters, and select Limit to genes, choosing with With Cleavage site (Signalp) from the drop-down and then Only. Clicking on Count should reveal that you have filtered the dataset down to 217 genes.
Click on Attributes and expand GENE. Select Gene name. Now click on Results. The first 10 results are displayed by default; Display all results by selecting ALL from the drop down menu.
The output will display the Ensembl gene ID, Ensembl Transcript ID and gene names of all proteins with a Signalp cleavage site on mouse chromosome 9. If you prefer, you can also export as an Excel sheet by using the Export all results to XLS option.
Exporting homologues with BioMart
Go to Ensembl’s BioMart. For a list of Ciona savignyi Ensembl genes, export the human orthologues:
ENSCSAVG00000000002, ENSCSAVG00000000003, ENSCSAVG00000000006, ENSCSAVG00000000007, ENSCSAVG00000000009, ENSCSAVG00000000011
Do all of these genes have a homologue in human?
-
Go to BioMart (you can find a shortcut in the navigation bar at the top of any Ensemblpage) and click New. Choose the Ensembl Genes database. Choose the Ciona savignyi genes (CSAV 2.0) dataset.
-
Click on Filters in the left panel. Expand the GENE. Enter the gene list in the Input external references ID list box. Gene stable ID(s) should be preselected.
-
Click on Attributes in the left panel. Select the Homologues attributes at the top of the page. Expand the GENE section. Deselect Gene stable ID version, Transcript stable ID and Transcript stable ID version. Expand the ORTHOLOGUES [F-J] section. Select Human gene stable ID.
-
Click Results. Select View: All rows as HTML.
All but ENSCSAVG00000000006 have a homologue in human.
Convert IDs using BioMart
BioMart is a very handy tool when you want to convert IDs from different databases. The following is a list of 29 IDs of human proteins from the NCBI RefSeq database:
NP_001218, NP_203125, NP_203124, NP_203126, NP_001007233, NP_150636, NP_150635, NP_001214, NP_150637, NP_150634, NP_150649, NP_001216, NP_116787, NP_001217, NP_127463, NP_001220, NP_004338, NP_004337, NP_116786, NP_036246, NP_116756, NP_116759, NP_001221, NP_203519, NP_001073594, NP_001219, NP_001073593, NP_203520, NP_203522
Use BioMart in Ensembl to generate a list that shows to which Ensembl gene IDs and to which gene names these RefSeq IDs correspond. Do these 29 transcripts correspond to 29 genes?
-
Go to BioMart. You can find a shortcut to the tool on any Ensembl page in the navigation bar at the top of the page. Click New in the top left-hand menu if you need to start a new query. Choose the Ensembl Genes database. Choose the Human genes dataset.
-
Click on Filters in the left panel. Expand the GENE section. Select Input external references ID list - RefSeq peptide ID(s) and enter the list of IDs in the text box (either comma separated or as a list).
HINT: You may have to scroll down the menu to see these.
Count shows 10 genes (remember one gene may have multiple splice variants coding for different proteins, that is the reason why these 29 proteins do not correspond to 29 genes).
-
Click on Attributes in the left panel. Select the Features attributes page. Expand the External section. Select HGNC symbol and RefSeq Peptide ID from the External References section.
-
Click the Results button on the toolbar. Select View: All rows as HTML or export all results to a file.
Export structural variants
You can use BioMart to query variants, not just genes. (Make sure you use the right Datasets.)
(a) Export the study accession, source name, chromosome, sequence region start and end (in bp) of human structural variations (SV) on chromosome 1, starting at 130,408 and ending at 210,597.
(b) In a new BioMart query, find the alleles, phenotype descriptions, and associated genes for the human SNPs rs566014072 and rs754099015. Can you view this same information in the Ensembl browser?
(a) Choose Ensembl Variation and Human Structural Variants (GRCh38).
Filters: Region: Chromosome 1, Base pair start: 130408, Base pair end: 210597
Count shows 87 structural variants.
Attributes: Structural Variation (SV) Information: DGVa Study Accession and Source Name, Structural Variation (SV) Location: Chromosome/scaffold name, Chromosome/scaffold position start (bp) and Chromosome/scaffold position end (bp).
(b) Choose Ensembl Variation and Human Short Variation (SNPs and indels) (GRCh38).
Filters: Filter by Variation name enter: rs566014072, rs754099015
Attributes: Variant Name, Variant Alleles, Phenotype description and Associated gene.
You can view this same information in the Ensembl browser. Click on one of the variation IDs (names) in the result table. The variation tab should open in the Ensembl browser. Click Phenotype Data.
Find genes associated with array probes
Forrest et al performed a microarray analysis of peripheral blood mononuclear cell gene expression in benzene-exposed workers (Environ Health Perspect. 2005 June; 113(6): 801–807). The microarray used was the human Affymetrix U133A/B (also called U133 plus 2) GeneChip. The top 25 up-regulated probe-sets were:
207630_s_at 221840_at 219228_at 204924_at 227613_at 223454_at 228962_at 214696_at 210732_s_at 212370_at 225390_s_at 227645_at 226652_at 221641_s_at 202055_at 226743_at 228393_s_at 225120_at 218515_at 202224_at 200614_at 212014_x_at 223461_at 209835_x_at 213315_x_at
(a) Retrieve for the genes corresponding to these probe-sets the Ensembl Gene and Transcript IDs as well as their HGNC symbols and descriptions.
(b) In order to analyse these genes for possible promoter/enhancer elements, retrieve the 2000 bp upstream of the transcripts of these genes.
(c) In order to be able to study these human genes in mouse, identify their mouse orthologues. Also retrieve the genomic coordinates of these orthologues.
(a) Click New. Choose the ENSEMBL Genes database. Choose the Human genes (GRCh38) dataset.
Click on Filters in the left panel. Expand the GENE section by clicking on the + box. Select Input microarray probes/probesets ID list - AFFY HG U133 Plus 2 probe ID(s) and enter the list of probeset IDs in the text box (either comma separated or as a list).
Count shows 26 genes match this list of probes.
Click on Attributes in the left panel. Select the Features attributes page. Expand the GENE section by clicking on the + box. In addition to the default selected attributes, select Description. Expand the External section by clicking on the + box. Select HGNC symbol from the External References section and AFFY HG U133 Plus 2 probe from the Microarray probes/probesets attributes section.
Click the Results button on the toolbar. Select View All rows as HTML or export all results to a file. Tick the box Unique results only.
Your results should show that the 25 probes map to 26 Ensembl genes.
(b) Don’t change Dataset and Filters – simply click on Attributes.
Select the Sequences attributes page. Expand the SEQUENCES section by clicking on the + box. Select Flank (Transcript) and enter 2000 in the Upstream flank text box. Expand the Header information section by clicking on the + box. Select, in addition to the default selected attributes, Gene description and Gene name.
Note: Flank (Transcript) will give the flanks for all transcripts of a gene with multiple transcripts. Flank (Gene) will give the flanks for one possible transcript in a gene (the most 5’ coordinates for upstream flanking).
Click the Results button on the toolbar.
(c) You can leave the Dataset and Filters the same, and go directly to the Attributes section:
Click on Attributes in the left panel. Select the Homologues attributes page. Expand the GENE section by clicking on the + box. Select Gene name. Deselect Ensembl Transcript ID. Expand the ORTHOLOGUES [K-O] section by clicking on the + box. Select Mouse gene stable ID, Mouse chromosome/scaffold name, Mouse chromosome/scaffold start (bp) and Mouse chromosome/scaffold end (bp).
Click the Results button on the toolbar. Select View All rows as HTML or export all results to a file.
Your results should show that for most of the human genes at least one mouse orthologue has been identified.
Exporting paralogues with BioMart
Export a list of all human genes on chromosome 14 which have a paralogue, including the gene names, the last common ancestor and the identity between the genes. How many genes on chromosome 14 have a paralogue?
Go to BioMart and click New. Choose the Ensembl Genes database. Choose the Human genes dataset.
Click on Filters in the left panel. Expand the REGION section by clicking on the + box and select Chromosome/scaffold – 14. Under MULTI SPECIES COMPARISONS select Homologue filters – Paralogous Human Genes: Only. Click the Count button in the side menu.
There are 806 genes on chromosome 14 which have a paralogue.
Click on Attributes in the left panel. Select Homologues from the six options at the top. Expand the GENE section by clicking on the + box. Deselect Transcript stable ID and Transcript stable ID version and select Gene name. Under PARALOGUES select Human paralogue gene stable ID, Human paralogue associated gene name, Paralogue last common ancestor with Human, Paralogue %id. target Human gene identical to query gene and Paralogue %id. query gene identical to target Human gene. Click the Results button on the toolbar. Select View: All rows as HTML or Export all results to a File.
Exporting regulatory features with BioMart
Using the Human Regulatory Features dataset, export a list of all enhancers falling in cytogenetic band q13.2 on chromosome 22 and their activity in Aorta. How many of them are active?
Go to BioMart and click New. Choose the Ensembl Regulation database. Choose the Human Regulatory Features dataset.
Click on Filters in the left panel. Expand the REGULATORY FEATURES section by clicking on the + box and select the following:
- Chromosome - 22
- Karyotype band: Band start – q13.2, Band end - q13.2
- Feature Type – Enhancer
- Epigenome name – aorta
Click on Attributes in the left panel. Select Chromosome/scaffold name, Start (bp), End (bp), Feature type, Regulatory stable ID, Activity and Epigenome name. Click the Results button to see the results table. Select View: All and choose to see Unique results only.
There is only one enhancer active in aorta in this cytogenetic band: ENSR00001239875.
Exporting histone modification sites with BioMart
Using the Human Regulatory Evidence dataset, export a list of all H3K9me3 modified loci on chromosome Y in Aorta. What is the source of this evidence?
Go to BioMart and click New. Choose the Ensembl Regulation database. Choose the Human Regulatory Evidence dataset.
Click on Filters in the left panel. Expand the REGULATORY EVIDENCE section by clicking on the + box and select Chromosome - Y, Feature Type – H3K9me3, and Epigenome – aorta.
Click on Attributes in the left panel. Select Chromosome/scaffold name, Start (bp), End (bp), Feature type, Epigenome name and Project name. Click the Results button to see the results table. Select View: All rows as HTML or Export all results to a File.
This data comes from the Roadmap Epigenomics.